---
author:
- name: Evelyn Metzger
orcid: 0000-0002-4074-9003
affiliations:
- ref: bsb
- ref: eveilyeverafter
execute:
eval: false
freeze: auto
message: true
warning: false
self-contained: false
code-fold: false
code-tools: true
code-annotations: hover
engine: knitr
prefer-html: true
format: live-html
resources:
- assets/interactives/ecdf.parquet
---
# Differential Expression {#sec-de}
```{r}
#| label: Preamble
#| eval: true
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
source("./preamble.R")
library(ggplot2)
library(stringr)
reticulate::source_python("./preamble.py")
analysis_dir <- file.path(getwd(), "analysis_results")
input_dir <- file.path(analysis_dir, "input_files")
output_dir <- file.path(analysis_dir, "output_files")
analysis_asset_dir <- "./assets/analysis_results" # <1>
ct_dir <- file.path(output_dir, "ct")
de_dir <- file.path(output_dir, "de")
if(!dir.exists(de_dir)){
dir.create(de_dir, recursive = TRUE)
}
results_list_file = file.path(analysis_dir, "results_list.rds")
if(!file.exists(results_list_file)){
results_list <- list()
saveRDS(results_list, results_list_file)
} else {
results_list <- readRDS(results_list_file)
}
library(smiDE)
```
## Introduction
There are several points during exploratory data analysis were observations
turn into biologicaly hypotheses. Often times we want to convert a given biological
hypothesis into a statistical one to formally test for differences between two
or more groups. Differential Expression (DE) analysis is one such tool that allows us
to test these hypotheses. In this chapter we'll run differential expression
analysis using the `smiDE` R package that was specifically designed to account
for the spatial relationship found in CosMx SMI data. We'll continue to work with
the colon cancer dataset where we previously
identified two tumor-rich spatial domains -- the Tumor Core and the Desmoplastic Stroma --
with varying levels of PROGENy pathway
enrichment scores.
There are many ways one could go about using smiDE and enumerating all the ways
is beyond the scope of this chapter. Let's consider two motivating questions centered
around the tumor cells themselves:
1) Location based: How are tumor cells' expression influenced by the domain in which they reside? Here the grouping variable will be the annotated domain a tumor cell belongs to.
2) "Immune Pressure" based: How are tumor cells' expression influenced by the number of spatially adjacent
immune cells? In this case, the grouping variable will be a numeric value (_e.g._, a given tumor cell might have 10 immune cells within a given spatial radius while another tumor cell might have zero immune cells nearby).
Most of this chapter will be based in R and so we'll first convert our
annData object before we tackle these questions. For each DE question there will
be a lot of results to look at and the `smiDE` package has ways to evaluate and explore
these results. Instead of enumerating all the volcano plots and other results here, I will
show how I loaded them into the
[`Differential Expression Explorer`](https://nanostring-biostats.github.io/blog-previews/pr-292/bbytes/de_explorer/index.html){target="_blank"} Browser Byte Dashboard.
## Converting Data
Read the annData object.
```{python}
#| label: load-anndata
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "python code"
if 'adata' not in dir():
filename = os.path.join(r.analysis_dir, "anndata-7-domain_annotations.h5ad")
adata = ad.read_h5ad(filename)
counts = adata.layers['counts'].astype('int64')
norm = adata.layers['TC']
```
Before we switch over to R for DE, let's calculate the number of immune cells within 80 microns of
a tumor cell. We'll use this information for the second DE question. We'll create
a KDtree of the immune cells and then query all of the cells in the annData object
to tally the number of adjacent immune cells within 80 microns.
```{python}
#| label: load-n_ct_immune_neighbors
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "python code"
from sklearn.neighbors import KDTree
import numpy as np
import pandas as pd
radius_mm = 0.080
cell_type_column = "celltype_broad"
immune_cell_types = [
"bcell", "dendritic", "macrophage", "mast",
"mix_NK_Lymphoid", "monocyte", "neutrophil", "nk", "plasma", "tcell"
]
coords = adata.obsm['spatial']
is_immune = adata.obs[cell_type_column].isin(immune_cell_types).values
immune_coords = coords[is_immune]
tree = KDTree(immune_coords)
# Query all cells
tally = tree.query_radius(coords, r=radius_mm, count_only=True)
# If the focal cell itself is immune, subtract 1 from the total
final_tally = tally - is_immune.astype(int)
adata.obs['n_ct_immune_neighbors'] = final_tally
```
Now switch to R and load and "munge" the data.
```{r}
#| label: py-to-R-objects
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
counts_mat <- Matrix::t(py$counts)
colnames(counts_mat) <- py$adata$obs_names$to_list()
rownames(counts_mat) <- py$adata$var_names$to_list()
norm_mat <- Matrix::t(py$norm)
colnames(norm_mat) <- py$adata$obs_names$to_list()
rownames(norm_mat) <- py$adata$var_names$to_list()
meta_df <- py$adata$obs
rownames(meta_df) <- meta_df$cell_id_numeric <- colnames(counts_mat)
totalcount_scalefactors <- mean(meta_df[["nCount_RNA"]]) / meta_df[["nCount_RNA"]]
names(totalcount_scalefactors) <- rownames(meta_df)
xy <- py$adata$obsm['spatial']
colnames(xy) <- c("sdimx", "sdimy")
meta_df <- cbind(meta_df, xy)
```
## Setup
There are a few cells that do not have a spatial domain assignment. These are
labeled as missing values which isn't allowed for smiDE. Therefore, subset the expression
matrices and metadata to include only cells with spatial domains.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
cells_use <- !is.na(meta_df$annotated_domain)
meta_use <- meta_df[cells_use,]
counts_mat_use <- counts_mat[,cells_use]
norm_mat_use <- norm_mat[,cells_use]
```
### Overlap Ratio Metric
We want to exclude any gene targets that might be from abutting cells that are not tumor
cells. For example, _JCHAIN_ is often highly expressed in plasma cells, so we might
see _JCHAIN_ spuriously show up as higher in tumor cells in a domain with a high
abundance of plasma cells. The function
below mitigates that effect by considering the gene expression of a focal cell
relative to close neighbors who are not of the same cell type. See the documentation [here](https://github.com/Nanostring-Biostats/CosMx-Analysis-Scratch-Space/tree/Main/_code/smiDE)
and the `Choosing an overlap ratio` deep-dive box below for more details.
We'll select genes that have a ratio of less than 1.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
overlap_metrics <- smiDE::overlap_ratio_metric(assay_matrix = norm_mat_use,
metadata = meta_use,
cellid_col = "cell_id_numeric",
cluster_col = "celltype_broad",
sdimx_col = "sdimx",
sdimy_col = "sdimy",
radius = 0.05
)
genes_to_analyze <- overlap_metrics[celltype_broad=="tumor"][ratio < 1][["target"]]
saveRDS(genes_to_analyze, file=file.path(de_dir, "genes_to_analyze.rds"))
```
:::{.noteworthybox title="Choosing an overlap ratio" collapse="true"}
We can define the **intrinsic expression** of a cell type as the collection of
gene targets demonstrating consistently higher expression within that focal cell
type compared to its immediate spatial neighbors. Because pervasive, uniform
gene translation across all cells is biologically improbable, spatial
differential expression (DE) models can account for this localized specificity.
To quantify this localized specificity, we establish an empirical **overlap
ratio** for each target-by-cell-type combination (e.g., Gene A in Cell Type B).
For a given gene, this statistic is calculated by taking its average expression
in non-focal cells and dividing it by its average expression in the focal
cell type.
Leveraging these local spatial patterns prior to DE analysis provides a
systematic way to:
* **Filter spurious signals:** Identify genes whose expression is primarily
bleeding over from adjacent cells.
* **Reduce the multiple testing burden:** Limit the number of hypotheses
(targets) we test to those with true biological relevance in the cells of interest.
Specifically, targets driven heavily by neighboring non-focal cells (such as
fibroblasts or NK cells) will yield a ratio > 1. For analytical clarity, this
overlap is typically evaluated in log space, where expression predominantly
originating from the non-focal neighborhood corresponds to a log2(ratio) > 0.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
overlap_metrics$log2ratio <- log2(overlap_metrics$ratio)
```
Let's explore how this transformed ratio behaves. We'll compute the empirical
cummulative distribution function (eCDF) for each focal cell type that is in the
`overlap_metrics` R object. We do this simply by grouping by cell type, sorting
the results, and then returning the proportion of targets that have a smaller
ratio.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
ecdf <- do.call(rbind, lapply(unique(overlap_metrics[['celltype_broad']]), function(fct){
x <- overlap_metrics[overlap_metrics[['celltype_broad']]==fct,]
x <- x[order(x[['log2ratio']]),]
return(
data.frame(focal_cell_type = x[['celltype_broad']],
target=x[['target']], log2ratio = x[['log2ratio']],
prop = (1:nrow(x)) / nrow(x))
)
}))
saveRDS(ecdf, file=file.path(de_dir, "ecdf.rds"))
```
And here is what those eCDFs look like.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
celltype_broad_colors <- results_list[['celltype_broad_colors']]
names(celltype_broad_colors) <- results_list[['celltype_broad_names']]
p <- ggplot() +
geom_line(data=ecdf,
aes(x=log2ratio, y=prop, colour=focal_cell_type, group=focal_cell_type)) +
theme_bw() +
scale_color_manual(values=celltype_broad_colors) +
xlab("log2 (Mean Expression in Neighbors of Focal Cell Type / Mean Expression in Focal Cell Type)") +
ylab("Empirical Cumulative Distribution Function") +
guides(colour=guide_legend(title="Cell Type"))
ggsave(p, filename=file.path(de_dir, "ecdfs.png"),
width=8, height=6, dpi=150, type="cairo")
```
```{r}
#| label: fig-de-ecdf
#| message: false
#| warning: false
#| echo: false
#| fig.width: 8
#| fig.height: 6
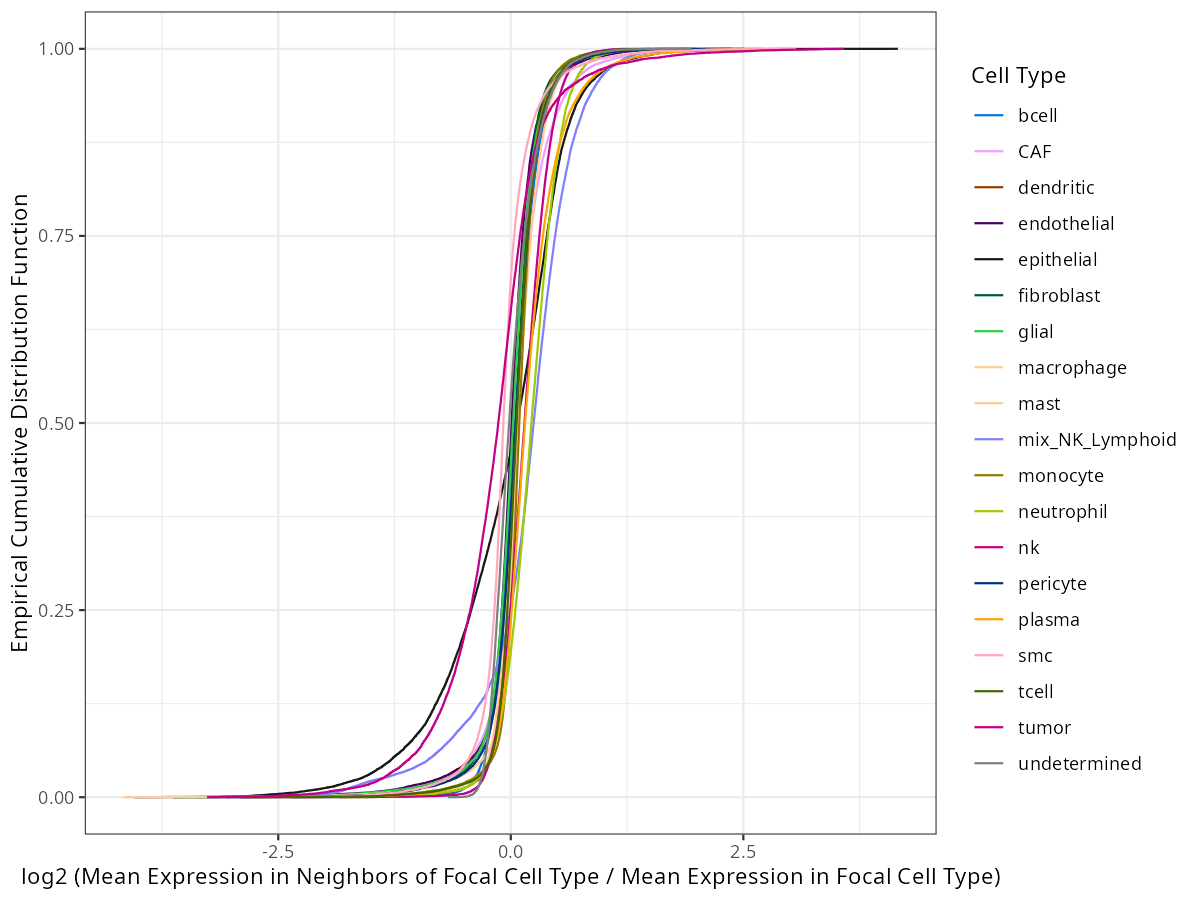
#| fig-cap: "Empirical cummulative distribution for each cell type. Y proportion of gene targets have a log2 ratio less than X. "
#| eval: true
render(file.path(analysis_asset_dir, "de"), "ecdfs.png", de_dir, overwrite=TRUE)
```
You can see from @fig-de-ecdf that the proportion of targets that are more abundant
in a given cell type relative to other cell types depends on the cell type. If we
look only at X = 0, we see the proportion of targets that have higher intrinsic
expression for each cell type. Those values are shown in table @tbl-de-ecdf.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
ecdf_at_zero <- ecdf %>%
group_by(focal_cell_type) %>%
summarize(
prop_at_zero = approx(x = log2ratio, y = prop, xout = 0)$y
) %>% arrange(prop_at_zero)
ecdf_at_zero$targets_below_zero <- round(ecdf_at_zero$prop_at_zero * length(unique(ecdf$target)))
saveRDS(ecdf_at_zero, file=file.path(de_dir, "ecdf_at_zero.rds"))
```
```{r}
#| label: tbl-de-ecdf
#| message: false
#| warning: false
#| echo: false
#| fig.width: 8
#| fig.height: 6
#| tbl-cap: "Approximate number of targets that have focal cell type expression that is greater than neighboring non-focal cell type expression."
#| eval: true
ecdf_at_zero <- readRDS(file.path(de_dir, "ecdf_at_zero.rds"))
ecdf_at_zero %>% knitr::kable(
col.names = c(
"Cell Type",
"Prop. Targets below log2(ratio) = 0",
"Total Targets below log2(ratio) = 0"
),
digits = 4, # Rounds the proportion to 4 decimal places
format.args = list(big.mark = ",") # Adds commas to large integers
) %>%
kable_styling(
bootstrap_options = c("striped", "hover", "condensed"),
full_width = FALSE,
position = "left"
)
```
For our cell type of interest, tumor, we see that the number of intrinsic expression is 12,303 targets.
And what about our intuition about _JCHAIN_ in tumor? The log2 ratio of _JCHAIN_ in tumor
is 2.02 (ratio = 4.07) and that is more extreme than 99% of targets. In plasma cells,
on the other hand, the log2 ratio of _JCHAIN_ is -2.80 (ratio = 0.14) which happens to be
the lowest ratio for plasma cells which makes biological sense.
```{r}
#| eval: true
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
ecdf <- readRDS(file.path(de_dir, "ecdf.rds"))
filter(ecdf, focal_cell_type %in% c("tumor", "plasma"), target=="JCHAIN")
```
In this chapter we chose a log2 ratio of < 0 (ratio < 1). In practice that threshold
is a good "rule of thumb"
but the choice is entirely up to the user. Here's an eCDF plot that focuses on
tumor only. The rug above the x-axis shows the relative
density of targets. Hover over the plot to more detail.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
ecdf_file <- "./assets/interactives/ecdf.parquet"
nanoparquet::write_parquet(ecdf %>% filter(focal_cell_type == "tumor") %>% select(-focal_cell_type), ecdf_file)
```
```{ojs}
//| echo: false
//| eval: true
// Initialize DuckDB and load data
db = DuckDBClient.of({
ecdf_data: FileAttachment("assets/interactives/ecdf.parquet")
})
// Query data from DuckDB
raw_data = db.sql`SELECT * FROM ecdf_data ORDER BY log2ratio ASC`
// Format data for Plot
plotData = raw_data.map((d, i) => ({
...d,
// Force numeric types to prevent "scale incompatible" errors
log2ratio: Number(d.log2ratio),
prop: Number(d.prop),
count_less: i
}))
// Render the eCDF Chart
chart = Plot.plot({
width: 800,
height: 500,
x: {
label: "Log2 Ratio",
type: "linear"
},
y: {
label: "Proportion",
type: "linear"
},
marks: [
// 1. The Binned Rug Plot (Optimized for 20k rows)
Plot.ruleX(plotData, Plot.binX(
{ strokeOpacity: "count" },
{
x: "log2ratio",
y1: 0, // Start at X-axis
y2: 0.03, // 3% tick height
strokeWidth: 3,
thresholds: 200
}
)),
// 2. The main eCDF line
Plot.line(plotData, {
x: "log2ratio",
y: "prop",
stroke: "#C11A7A",
strokeWidth: 2.5
}),
// 3. The constrained vertical drop-line on hover
Plot.ruleX(plotData, Plot.pointerX({
x: "log2ratio",
y1: 0,
y2: "prop",
stroke: "gray",
strokeDasharray: "4,4",
strokeWidth: 1.5
})),
// 4. The Hover Tooltip (with rounded formatting)
Plot.tip(plotData, Plot.pointerX({
x: "log2ratio",
y: "prop",
// Using .toFixed() and .toPrecision() to format the long decimals
title: d => `Target: ${d.target}\nTargets < Y: ${d.count_less}\nLog2 Ratio: ${d.log2ratio.toFixed(4)}\nProportion: ${d.prop.toPrecision(3)}`
})),
// 5. Table Selection Highlights
Plot.dot(selected_targets, {
x: "log2ratio",
y: "prop",
fill: "red",
r: 5
}),
Plot.text(selected_targets, {
x: "log2ratio",
y: "prop",
text: "target",
dy: -12,
fill: "red",
fontWeight: "bold"
})
]
})
```
If you are interested in a particular gene, you can search it in the table below.
Selecting a gene will label it on the figure above.
```{ojs}
//| echo: false
//| eval: true
viewof selected_targets = Inputs.table(plotData, {
multiple: true,
required: false,
columns: ["target", "log2ratio", "prop"],
header: {
target: "Target",
log2ratio: "Log2 Ratio",
prop: "Proportion"
}
})
```
:::
### Compute cell-cell adjacencies
This code computes a table identifying the neighbor cells of each cell within
0.05 mm, and records the corresponding celltype ("celltype_broad") of each neighbor.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
pre_de_obj <- pre_de(metadata = meta_df
,cell_type_metadata_colname = "celltype_broad"
,mm_radius = 0.05
,sdimx_colname = "sdimx"
,sdimy_colname = "sdimy"
,verbose=FALSE
,cellid_colname = "cell_id_numeric"
)
saveRDS(pre_de_obj, file=file.path(de_dir, "prede_obj.rds"))
```
## DE1: Amongst Domains
Let's run DE on tumor cells betwen each pairwise spatial domain. This section
shows you how to create the DE analysis, run it, and then package up the results
to view in an interactive dashboard.
:::{.callout-note}
If you want to skip ahead to see the results
in the dashboard, they are already available. This particular result is the study
that is labeled "Tumor differences between domains".
:::
In the block below, we'll focus on the tumor cells within `celltype_broad` and run differential expression
analysis between all pairwise combinations of `annotated_domain` on the filtered
list of gene targets that we identified above.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
meta_use <- meta_df %>% filter(celltype_broad == "tumor")
assay_matrix_use <- counts_mat[,meta_use$cell_id_numeric]
tc_scalefactors <- mean(meta_df$nCount_RNA)/meta_df$nCount_RNA
names(tc_scalefactors) <- meta_df$cell_id_numeric
de_obj <-
smi_de(assay_matrix = assay_matrix_use
,metadata = meta_use
,formula = ~RankNorm(otherct_expr) + annotated_domain + offset(log(nCount_RNA))
,pre_de_obj = pre_de_obj
,neighbor_expr_cell_type_metadata_colname = "celltype_broad"
,neighbor_expr_overlap_weight_colname = NULL
,neighbor_expr_overlap_agg ="sum"
,neighbor_expr_totalcount_normalize = TRUE
,neighbor_expr_totalcount_scalefactor = tc_scalefactors
,family="nbinom2"
,cellid_colname = "cell_id_numeric"
,targets=genes_to_analyze,
nCores=30
)
saveRDS(de_obj, file=file.path(de_dir, "de1_obj.rds"))
```
### Preparing Results for Interactive Dashboard
We'll follow the instructions on the [dashboard](https://nanostring-biostats.github.io/blog-previews/pr-292/bbytes/de_explorer/index.html#sec-convert-smide)
to convert the `de_obj` to the format expected by the Differential Expression Explorer.
Specifically, we are going to make the following datasets available in a parquet format:
1. Study header: simple description of the DE
2. Pairwise data: a data frame containing the individual pairwoise contrasts
3. Marginal data: the maringal means
4. Cell metadata: cell metadata columns used in the formula + coordinates
For the study header, this is simply was way to jot down a description of the
DE itself so that it can be distinugished from other results on the dashboard
that are either publicaly available or that you loaded.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
study_header <- data.frame(
`Name` = "Tumor cells across spatial domains",
`Description` = "Colon Cancer WTX sample",
`Formala` = "~RankNorm(otherct_expr) + annotated_domain + offset(log(nCount_RNA))",
`Targets tested` = length(genes_to_analyze)
)
```
Since the output structure differs slighlty depending on whether your DE results are based on discrete or continous terms, we’ll use this harmonizing function to format the results into a single format that the dashboard can understand. Specifically, the function below converts the pairwise results and the emmeans results.
```{r}
library(data.table)
library(dplyr)
library(stringr)
#' Harmonize smiDE Results for Dashboard
#'
#' @param res_list The output list directly from smiDE::results(de_obj)
#' @return A list containing 'pairwise' and 'emmeans' data.frames with synchronized continuous labels
harmonize_de_results <- function(res_list) {
emmeans_raw <- res_list$emmeans
if ("term" %in% names(emmeans_raw)) {
emmeans_raw <- emmeans_raw[term != "otherct_expr"]
}
emmeans_processed <- emmeans_raw %>%
mutate(
# Extract numeric suffix from level (e.g., "term42.697" -> "42.697")
suffix_str = str_remove(level, fixed(term)),
suffix_val = suppressWarnings(as.numeric(suffix_str)),
is_continuous = !is.na(suffix_val) & str_starts(level, fixed(term))
) %>%
mutate(
clean_level = case_when(
is_continuous ~ {
direction <- case_when(
category == "c1" ~ "+ 1SD",
category == "c2" ~ "",
TRUE ~ ""
)
paste0("mean ", direction, " (", sprintf("%.2f", suffix_val), ")")
},
TRUE ~ level
),
lookup_key = ifelse(is_continuous,
paste0(term, "_", sprintf("%.2f", suffix_val)),
NA_character_)
)
continuous_map <- emmeans_processed %>%
filter(is_continuous) %>%
select(lookup_key, clean_level) %>%
distinct() %>%
tibble::deframe()
emmeans_out <- emmeans_processed %>%
select(term, level = clean_level, target, response, asymp.LCL, asymp.UCL) %>%
as.data.frame()
pairwise_combined <- rbindlist(
res_list[c("pairwise", "one.vs.rest", "one.vs.all")],
idcol = "contrast_type",
use.names = TRUE,
fill = TRUE
)
if ("term" %in% names(pairwise_combined)) {
pairwise_combined <- pairwise_combined[term != "otherct_expr"]
}
if (!"ncells_1" %in% names(pairwise_combined)) pairwise_combined[, ncells_1 := NA]
if (!"ncells_2" %in% names(pairwise_combined)) pairwise_combined[, ncells_2 := NA]
pairwise_out <- pairwise_combined %>%
mutate(
log2fc = log2(fold_change),
is_continuous_row = is.na(ncells_1) & is.na(ncells_2)
) %>%
rowwise() %>%
mutate(
contrast = if (is_continuous_row) {
parts <- str_split(contrast, " / ", simplify = TRUE)
val1 <- as.numeric(str_remove(parts[1], fixed(term)))
val2 <- as.numeric(str_remove(parts[2], fixed(term)))
key1 <- paste0(term, "_", sprintf("%.2f", val1))
key2 <- paste0(term, "_", sprintf("%.2f", val2))
label1 <- ifelse(key1 %in% names(continuous_map), continuous_map[key1], parts[1])
label2 <- ifelse(key2 %in% names(continuous_map), continuous_map[key2], parts[2])
paste0(label1, " / ", label2)
} else {
contrast
}
) %>%
ungroup() %>%
select(term, contrast, target, log2fc, p.value, ncells_1, ncells_2) %>%
as.data.frame()
return(list(pairwise = pairwise_out, emmeans = emmeans_out))
}
```
And now we harmonize the results after extracting them from the `de_obj` object.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
res <- results(de_obj)
res_formatted <- harmonize_de_results(res)
pairwise <- res_formatted$pairwise
emmeans <- res_formatted$emmeans
```
I often like to plot the cells that were used in my grouping. Let's extract the spatial coordinates, the
slide identifier, the
annotated domain, and the cell type column from the metadata.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
meta_display <- data.table(meta_df)
meta_display <- meta_display[, .(
x_slide_mm = sdimx,
y_slide_mm = sdimy,
celltype_broad,
annotated_domain
)]
if('slide_id_numeric' %in% colnames(meta_df)){
meta_display$slide_id_numeric <- meta_df$slide_id_numeric
} else {
meta_display$slide_id_numeric <- 1L
}
```
Alternatively, if you just want the volcano plots and emmeans to show on the dashboard
and wanted to skip the spatial plotting altogether, I would just create and pass a placeholder data.frame like so:
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
create_mock_sp_data <- function(emmeans_formatted) {
terms <- emmeans_formatted %>%
select(term) %>%
pull()
mock_template <- data.frame(
'x_slide_mm' = c(0, 0, 1, 1),
'y_slide_mm' = c(0, 1, 0, 1),
'slide_id_numeric' = rep(1L, 4)
)
for(i in seq_len(length(terms))){
mock_template[[terms[i]]] <- rep(NA, 4)
}
return(mock_template)
}
# this will just add 4 rows data which the dashboard uses as a signature for
# "Not Applicable"
meta_display <- create_mock_sp_data(emmeans)
```
At this point we are ready to package these four components so they can be loaded
in the dashboard. The two steps to this procedure is:
1. convert data to parquet.
2. zip up all parquet files into a single zip file.
When converting data to parquet, we’ll use this write_opt_parquet function. It’s just a wrapper function for writing to parquet but, to save memory, one could reduce the numeric precision of columns from R’s float64 to either 32 bit or 16 bit, if desired. Keep in mind that things like p-values will likely need greater precision.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
library(arrow)
write_opt_parquet <- function(df, filename, f32_cols = NULL, f16_cols = NULL) {
# Convert to Arrow Table
arrow_table <- as_arrow_table(df)
# Cast Float32 columns
if (!is.null(f32_cols)) {
# Check which requested columns actually exist in this dataframe
valid_cols <- intersect(names(df), f32_cols)
if(length(valid_cols) > 0) {
arrow_table <- arrow_table %>%
mutate(across(all_of(valid_cols), ~ cast(.x, float32())))
}
}
# Cast Float16 columns
if (!is.null(f16_cols)) {
valid_cols <- intersect(names(df), f16_cols)
if(length(valid_cols) > 0) {
arrow_table <- arrow_table %>%
mutate(across(all_of(valid_cols), ~ cast(.x, float16())))
}
}
# Write to disk
write_parquet(arrow_table, filename)
}
```
Write the parquet files to disk. Note that the folder names can vary but the file names must match exactly.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
parquet_dir <- file.path(de_dir, "de1") # or whatever you like
dir.create(parquet_dir)
write_opt_parquet(study_header, file.path(parquet_dir, "study_header.parquet"))
write_opt_parquet(
pairwise,
file.path(parquet_dir, "pairwise.parquet")
)
write_opt_parquet(
emmeans,
file.path(parquet_dir, "emmeans.parquet")
)
write_opt_parquet(
meta_display,
file.path(parquet_dir, "cell_metadata.parquet"),
f32_cols = c('x_slide_mm', 'y_slide_mm'),
f16_cols = NULL
)
```
And finally, zip it up! You can name the zip file whatever you like but the parquet
file names themselves should not be adjusted.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
files_to_zip <- c("study_header.parquet",
"pairwise.parquet",
"emmeans.parquet",
"cell_metadata.parquet")
zip::zip(
zipfile = "you_can_name_this_whatever_you_like.zip",
files = files_to_zip,
root = parquet_dir
)
```
And that's it. If you are creating results on your own study, you could take your
zip file and load it into the dashboard. In this specific case, I loaded those
parquet files onto a remote repository so that they can be queried and available
to look at directly in the dashboard.
## DE 2: Tumors cells by number of immune neighbors
And now let's run DE on tumor cells with varying number of close immune cells.
The DE set up is slightly different than the first DE run but the rest of the
results -- extraction and packaging -- is the same as above.
:::{.callout-note}
To see this DE result on the dashboard, select the "Tumor by immune pressure" study.
:::
In the DE set up, we just switch out the grouping variable from `annotated_domain`
to `n_ct_immune_neighbors` that we computed above.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
tc_scalefactors <- mean(meta_df$nCount_RNA)/meta_df$nCount_RNA
names(tc_scalefactors) <- meta_df$cell_id_numeric
de2_obj <-
smi_de(assay_matrix = counts_mat
,metadata = meta_df
,formula = ~RankNorm(otherct_expr) + n_ct_immune_neighbors + offset(log(nCount_RNA))
,pre_de_obj = pre_de_obj
,neighbor_expr_cell_type_metadata_colname = "celltype_broad"
,neighbor_expr_overlap_weight_colname = NULL
,neighbor_expr_overlap_agg ="sum"
,neighbor_expr_totalcount_normalize = TRUE
,neighbor_expr_totalcount_scalefactor = tc_scalefactors
,family="nbinom2"
,cellid_colname = "cell_id_numeric"
,targets=genes_to_analyze,
nCores=10
)
saveRDS(de2_obj, file=file.path(de_dir, "de2_obj.rds"))
```
As with the first DE analysis, convert the data to the parquet files.
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
study_header2 <- data.frame(
`Name` = "Tumor cell expression by immunce pressure",
`Description` = "Colon Cancer WTX sample looking at tumor cell expression as a function of the number of immune cells within 80 µm.",
`Formala` = "~RankNorm(otherct_expr) + n_ct_immune_neighbors + offset(log(nCount_RNA))",
`Targets tested` = length(genes_to_analyze)
)
meta_display2 <- data.table(meta_df)
meta_display2 <- meta_display2[, .(
x_slide_mm = sdimx,
y_slide_mm = sdimy,
n_ct_immune_neighbors,
celltype_broad,
annotated_domain
)]
if('slide_id_numeric' %in% colnames(meta_df)){
meta_display2$slide_id_numeric <- meta_df$slide_id_numeric
} else {
meta_display2$slide_id_numeric <- 1L
}
res2 <- results(de2_obj)
res2_formatted <- harmonize_de_results(res2)
pairwise2 <- res2_formatted$pairwise
emmeans2 <- res2_formatted$emmeans
```
```{r}
#| eval: false
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
dir.create(file.path(de_dir, "de2"))
write_opt_parquet(
pairwise2,
file.path(de_dir, "de2", "pairwise.parquet")
)
write_opt_parquet(
emmeans2,
file.path(de_dir, "de2", "emmeans.parquet")
)
write_opt_parquet(
meta_display2,
file.path(de_dir, "de2", "cell_metadata.parquet"),
f32_cols = c('x_slide_mm', 'y_slide_mm'),
f16_cols = NULL
)
target_dir <- file.path(de_dir, "de2")
files_to_zip <- c("study_header.parquet",
"pairwise.parquet",
"emmeans.parquet",
"cell_metadata.parquet",
'overlap.parquet')
zip::zip(
zipfile = "de2.zip",
files = files_to_zip,
root = target_dir
)
```
## Examine the results
You can explore the results form this chapter interactively in the Scratch
Space [Differential Expression Explorer Browser Byte](https://nanostring-biostats.github.io/CosMx-Analysis-Scratch-Space/bbytes/de_explorer/){target="_blank"}. This
will include volcano plots, tables, and more.