---
subtitle: Classifying cells into types
author:
- name: Evelyn Metzger
orcid: 0000-0002-4074-9003
affiliations:
- ref: bsb
- ref: eveilyeverafter
execute:
eval: false
freeze: auto
message: true
warning: false
self-contained: false
code-fold: false
code-tools: true
code-annotations: hover
engine: knitr
prefer-html: true
format: live-html
resources:
- ./assets/analysis_results/ct
---
{{< include ./_extensions/r-wasm/live/_knitr.qmd >}}
# Cell Typing {#sec-cell-typing}
Okay, let's chart the course for this crucial phase of our analysis expedition:
cell typing. This is where we assign identities to the cell clusters discovered
in the previous chapter, transforming abstract groups into recognizable
biological entities like T cells, macrophages, or tumor cells.
If pre-processing was about plotting our trajectory through high-dimensional
space, cell typing is akin to navigating a dense asteroid field. It's a critical
step, but one fraught with potential obstacles. Assigning definitive labels can
be challenging, often requiring careful consideration of marker genes and
existing biological knowledge. What constitutes a "correct" cell type label can
even be subjective, depending entirely on the resolution needed for your
specific biological questions. Are you aiming to distinguish broad categories
like "immune" versus "tumor," or do you need to pinpoint specific T cell
subtypes? Like adjusting a microscope's focus, the level of detail required
dictates the approach. Navigating this field requires careful maneuvering and
the right instruments. Autopilot hasn't been invented just yet and so the best
recommendation need manual input.
One common hazard is attempting to directly apply analysis pipelines designed
for spatially-dissociated scRNA-seq. While scRNA-seq
vignettes offer valuable starting points, CosMx SMI data, generated by direct _in
situ_ hybridization, has fundamentally different characteristics. Simply copy-pasting
scRNA-seq methods without adaptation can lead to suboptimal or incomplete cell
type assignments. Instead, we must employ or adapt methods that leverage the
strengths of high-plex, direct hybridization data to achieve reliable
annotations. In doing so it's possible to achieve the most comprehensive and biologically-rich cell typing and sub-typing possible in spatial transcriptomics.
It's also crucial to remember that cell typing, while essential, isn't the final
destination of our journey. It's a powerful tool for organizing our cellular
map, allowing us to ask spatially-aware questions. However, other organizational
frameworks, such as classifying cells based on their spatial neighborhood or
"niche," are equally important for understanding the tissue's complex ecosystem.
Ultimately, cell typing is foundational: it establishes the structural anatomy
of the dataset, paving the way for us to investigate its functional physiology
in the next chapters.
In this chapter, we will navigate the cell typing process in the colon
dataset. The main goal is to generate cell type labels that are "close" while
also being approachable. I'll begin (@sec-ct-approaches) by surveying some of
the spatially unaware methods and then summarize the state of the direct
hybridization-inspired approaches. Then I will guide you through the specific,
hybrid workflow used for this colon dataset. We will start with the broad Leiden
clusters defined in the previous chapter, refine them based on spatial
observations, and assign primary identities using marker gene analysis. Finally,
for the more complex immune populations where standard clustering often
struggles, we will employ HieraType, a probabilistic method explicitly designed
to handle the specific characteristics of CosMx SMI data.
```{r}
#| label: Preamble
#| eval: true
#| echo: true
#| message: false
#| code-fold: true
#| code-summary: "R code"
source("./preamble.R")
reticulate::source_python("./preamble.py")
analysis_dir <- file.path(getwd(), "analysis_results")
input_dir <- file.path(analysis_dir, "input_files")
output_dir <- file.path(analysis_dir, "output_files")
analysis_asset_dir <- "./assets/analysis_results" # <1>
ct_dir <- file.path(output_dir, "ct")
if(!dir.exists(ct_dir)){
dir.create(ct_dir, recursive = TRUE)
}
results_list_file = file.path(analysis_dir, "results_list.rds")
if(!file.exists(results_list_file)){
results_list <- list()
saveRDS(results_list, results_list_file)
} else {
results_list <- readRDS(results_list_file)
}
library(scales)
library(Matrix)
source("./helpers.R")
```
## Cell Typing Approaches {#sec-ct-approaches}
### Non-spatial Cell Typing Approaches
Before discussing spatially-aware or technology-specific approaches, let's
briefly cover common methods designed for non-spatial, single-cell RNA sequencing analysis.
These techniques primarily leverage the gene expression matrix (`adata.X` and
normalized layers) and the cluster assignments (e.g., `adata.obs['leiden']`)
derived in the previous chapter. Many excellent vignettes exist for these
methods in popular frameworks like scanpy (Python) and Seurat (R), providing a
rough "10,000-foot view" of the data's cellular composition.
**Cluster Marker Gene Identification (Leiden/Louvain):** This is often a
foundational step in scRNA-seq. After obtaining clusters (like the Leiden clusters from
@sec-preprocessing), differential expression tests are performed between each
cluster and all other cells (or between pairs of clusters). This identifies
genes significantly upregulated within a specific cluster. Tools like
scanpy's `rank_genes_groups` or Seurat's `FindMarkers` are standard. The resulting
lists of marker genes are then manually compared against known canonical markers
from literature or cell atlases to assign putative cell type identities (e.g.,
observing high _CD3E_, _CD8A_ suggests a T cell cluster; high _EPCAM_, _KRT19_ suggests
epithelial cells). While effective, this relies heavily on prior biological
knowledge and the quality of the initial clustering. It also assumes that transcripts
found in the cell do not arise from any spatial co-localization or imperfect
cell-cell segmentation boundaries that can arise when assigning transcripts to cells.
**Nested / Hierarchical Clustering Analysis:** Sometimes, broad initial clusters
(like "Immune Cells" or "Fibroblasts") contain sub-populations that are meaningful
for your analysis. A
common refinement is then to subset the data to include only cells from a specific
Leiden cluster (or set of clusters) and then re-run the clustering
process within that subset. Identifying marker
genes for these sub-clusters can reveal finer cell types or states (_e.g._,
distinguishing CD4+ from CD8+ T cells, or identifying different fibroblast
subtypes. This iterative approach allows for a hierarchical understanding of
cell identity; however, the imperfect segmentation noted above is amplified in
this approach. Consider a workflow where one uses HVGs to create an initial set of
"Elementary" clusters and uses a marker gene-based approach to label these
broad clusters. When subsetting down to, say, fibroblasts and rerunning the above
HVG -> Leiden -> marker gene workflow, one may get non-fibroblast marker genes returned.
The reason for this is twofold. First the general fibroblast marker genes are no
longer as differentially expressed in the subset of Fibroblasts as they were in the full data
and 2) if there are cell segmentation errors of fibroblast sub-populations that are
spatially-associated with unrelated cell types -- like plasma cells -- the HVG
algorithm may identify spurious gene markers as highly variable. The result would
be markers like _JCHAIN_ in fibroblasts that happen to be located near a germinal
center, tertiary lymphoid structure, or similar. Thus, while this nested approach
can be valuable, it should be used with caution. For more information on this overlap
concept, see Dan McGuire's [blog post](https://nanostring-biostats.github.io/CosMx-Analysis-Scratch-Space/posts/smiDE/){target="_blank"} on smiDE.
**Reference-Based Annotation:** Instead of relying solely on _de novo_ marker finding,
many methods leverage existing annotated single-cell datasets (atlases) as
references. These algorithms typically project the query data
onto the reference or use statistical models trained on the reference to
directly assign cell type labels to your individual cells or clusters. Popular
tools include:
- [`scanpy.tl.ingest`](https://scanpy.readthedocs.io/en/stable/tutorials/basics/integrating-data-using-ingest.html):
Integrates query data onto an existing annotated AnnData object.
- Seurat's [`FindTransferAnchors`](https://satijalab.org/seurat/reference/findtransferanchors)
and [`TransferData`](https://satijalab.org/seurat/reference/transferdata): A widely used method in the R
ecosystem.
- InSituType's fully supervised method: InSituType -- while designed for spatial
data -- can be used as a fully supervised classifier. I'm including it here since
many reference datasets and atlases at the time of writing are based on scRNA-seq
data.
### Spatially-Informed and Hybridization-Aware Methods
Unlike standard scRNA-seq workflows, these approaches leverage the unique features of _in situ_ data: specifically the spatial coordinates, the negative probe background, and the available protein data.
**Background-Aware Modeling**. Methods like `InSituType` explicitly model the background noise using the negative control probes. This allows for more accurate classification of cells with low transcript counts that might be discarded or misclassified by standard scRNA-seq tools
**Spatial-Molecular Joint Inference**. Tools like [Banksy](https://www.nature.com/articles/s41588-024-01664-3) utilize the expression of a cell's physical neighbors to inform its identity. This leverages the biological reality that cells of the same type often cluster spatially, helping to smooth out technical noise in individual cells.
**Multi-modal Integration**. CosMx SMI often includes protein markers (e.g., CD45, PanCK) in the form of IF markers or -- more recently -- with true RNA and protein same-slide multiomics. "Dual-mode" typing strategies use these robust protein signals to define major lineages (e.g., "This cell is definitely CD45+ Immune") before using the RNA data to determine the specific subtype (e.g., "It is a CD8+ T Cell"). Similarly, [HieraType](https://nanostring-biostats.github.io/CosMx-Analysis-Scratch-Space/posts/multiomics-hieratype/#introduction)
is a new R package for supervised classification that can combine multi-omics
information to cell type. HieraType also classifies RNA-only data (as we'll see below).
**Reference-Based Annotation:** Reference profiles based on CosMx SMI can be used to
more quickly cell type new datasets. This approach can be much faster than cell
typing _de novo_ but requires such annotations to be available.
## Cell Type Determination
Just like there are many paths through an asteroid field, there are many ways to
navigate cell typing. I find the following approach makes a good starting place
for any analysis as it's relatively fast while still allowing for human intervention.
1. Use Leiden clusters identified in @sec-preprocessing as the basis.
1.1. _Optional_: refine Leiden clusters based on discovered biology. For example,
there may be some Leiden clusters that makes sense to merge together or some evidence
that splitting a Leiden cluster into two or splitting a given
Leiden cluster into multiple sub-clusters based on the spatial layout of cells.
2. Run HieraType to classify cells and use its integration function to merge its
supervised classifications with the unsupervised Leiden clusters.
3. Of the remaining unclassified cells, use marker genes to infer the most likely
cell type.
### Step 1: Read Leiden clusters
Load the data and make a copy of the `leiden_pr` metadata column that we'll use.
```{python}
#| label: load-anndata
#| eval: false
#| code-summary: "Python Code"
#| message: true
#| warning: false
#| code-fold: show
adata = ad.read_h5ad(os.path.join(r.analysis_dir, "anndata-2-leiden.h5ad"))
cluster_renaming = {}
initial = "C"
for name in adata.obs['leiden_pr'].unique():
cluster_renaming[name] = f"{initial}-{name}"
adata.obs['leiden'] = adata.obs['leiden_pr'].map(cluster_renaming)
```
### Step 2: Run HieraType
Install the HieraType package (and remotes), if needed. Then extract the lists
of marker genes.
```{r}
#| label: H-0
#| eval: false
#| code-summary: "R Code"
#| message: true
#| warning: false
#| code-fold: show
install.packages("mvtnorm")
remotes::install_github("Nanostring-Biostats/CosMx-Analysis-Scratch-Space",
subdir = "_code/HieraType", ref = "Main")
library(HieraType)
markerg <- unlist(lapply(c(
HieraType::markerslist_l1
,HieraType::markerslist_immune
,HieraType::markerslist_cd8tminor
,HieraType::markerslist_cd4tminor
,HieraType::markerslist_tcellmajor
), "[[", "predictors"))
marker_genes <- as.character(markerg)
```
Convert data to R.
```{python}
#| label: H-1
#| eval: false
#| code-summary: "Python Code"
#| message: false
#| warning: false
#| code-fold: show
total_counts_per_cell = adata.layers['counts'].sum(axis=1)
total_counts_per_cell = np.asarray(total_counts_per_cell).flatten()
total_counts_per_target = adata.layers['counts'].sum(axis=0)
total_counts_per_target = np.asarray(total_counts_per_target).flatten()
target_frequencies = total_counts_per_target / total_counts_per_target.sum()
# Include all HVGs and marker genes (even if not in the HVGs)
marker_genes = r.marker_genes
marker_mask = adata.var.index.isin(marker_genes)
highly_variable_mask = adata.var['highly_variable'].to_numpy()
adata.var['hieratype_target'] = (highly_variable_mask | marker_mask)
adata_ht = adata[:, adata.var.hieratype_target].copy()
ht_counts = adata_ht.layers['counts'].copy().astype(np.int64) # <1>
```
1. In order to use this matrix in R, we need to convert the 32 bit integer counts into 64 bit integers.
Follow [this tutorial](https://nanostring-biostats.github.io/CosMx-Analysis-Scratch-Space/posts/multiomics-hieratype/#rnaonly) to refine Leiden clusters with immune cells.
```{r}
#| label: H-2
#| eval: false
#| code-summary: "R Code"
#| message: true
#| warning: false
#| code-fold: show
ht_counts <- py$ht_counts
rownames(ht_counts) <- py$adata_ht$obs_names$to_list()
colnames(ht_counts) <- py$adata_ht$var_names$to_list()
ht_counts <- as(ht_counts, "CsparseMatrix")
pca_embeddings <- py$adata_ht$obsm['X_pca_pr'] # n_cells x n_pcs
target_frequencies <- py$target_frequencies
names(target_frequencies) <- py$adata$var_names$to_list()
target_frequencies <- target_frequencies[colnames(ht_counts)]
total_counts_per_cell <- py$total_counts_per_cell
# Assumes this was based on cosine.
sim_graph <- py$adata_ht$obsp['neighbors_pr_connectivities']
sim_graph <- as(sim_graph, "CsparseMatrix")
rownames(sim_graph) <- colnames(sim_graph) <- rownames(ht_counts)
```
Run the pipeline.
```{r}
#| label: H-3
#| eval: false
#| code-summary: "R Code"
#| message: true
#| warning: false
#| code-fold: show
pipeline <- HieraType::make_pipeline(
markerslists = list(
"l1" = HieraType::markerslist_multiomic_l1
,"l2" = HieraType::markerslist_multiomic_immune
,"lt" = HieraType::markerslist_multiomic_tcellmajor
,"lt4minor" = HieraType::markerslist_multiomic_cd4tminor
,"lt8minor" = HieraType::markerslist_multiomic_cd8tminor
)
,priors = list(
"l2" = "l1"
,"lt" = "l2"
,"lt4minor" = "lt"
,"lt8minor" = "lt"
)
,priors_category = list(
"l2" = "immune"
,"lt" = "tcell"
,"lt4minor" = "cd4t"
,"lt8minor" = "cd8t"
)
)
rnactobj <- HieraType::run_pipeline(
pipeline = pipeline,
counts_matrix = ht_counts,
adjacency_matrix = sim_graph[rownames(ht_counts), rownames(ht_counts)],
totalcounts = total_counts_per_cell,
gene_wise_frequency = target_frequencies
)
saveRDS(rnactobj, file=file.path(ct_dir, "rnactobj.rds"))
```
Merge the Leiden cluster calls with the HieraType
results. There are several ways we could do this but for this tutorial we'll
use HieraType's built-in `celltype_label_integration` function using the default
parameters. `celltype_label_integration` integrates the immune celltype annotations
from HieraType with unsupervised cluster labels. The goal of this step is to enable
granular detection of novel or tissue-specific cell types (unsupervised), along
with well known immune cell types (HieraType). The logic for merging these
annotations works like this:
1. All cells which are annotated with an immune-celltype HieraType label keep
their label.
2. All cells with a non-immune HieraType label take the unsupervised cluster label.
3. Unsupervised clusters that had a high rate of cells taking immune labels
(say 90%), and make up only a small proportion of all cells (say 1%) after step
1 are 'dissolved'. These dissoved cells are assigned the most common cluster label
(could be immune or unsupervised) amongst most the similar neighboring cells.
Here we used the neighbor cells from the adjacency matrix that was used to run HieraType.
```{r}
#| label: H-4
#| eval: false
#| code-summary: "R Code"
#| message: true
#| warning: false
#| code-fold: show
cell_type_dt <- rnactobj$post_probs$l1
cell_type_dt <- rename(cell_type_dt, cell_id = cell_ID, # <1>
Hieratype_celltype_fine = celltype_granular)
to_merge <- py$adata$obs %>% select(leiden)
cell_type_dt$Leiden <- to_merge$leiden[match(rownames(to_merge), cell_type_dt$cell_id)]
cell_type_dt <- celltype_label_integration(
metadata = cell_type_dt,
adjacency_mat = sim_graph[rownames(ht_counts), rownames(ht_counts)],
cellid_colname = 'cell_id',
supervised_colname = 'Hieratype_celltype_fine',
unsupervised_colname = 'Leiden')
cell_type_dt <- rename(cell_type_dt, merged = celltype)
saveRDS(cell_type_dt, file=file.path(ct_dir, "cell_type_dt.rds"))
```
1. cell_id here is the row name from the anndata object (as opposed to the c_i_j_k cell_ID notation that is sometimes used).
Save the new calumns back to the anndata object.
```{python}
#| label: H-5
#| eval: false
#| code-summary: "Python Code"
#| message: true
#| warning: false
#| code-fold: show
cell_type_df = r.cell_type_dt
adata.obs = adata.obs.join(cell_type_df[['cell_id', 'Hieratype_celltype_fine', 'merged']].set_index('cell_id'))
```
### Step 3: Classify remaining cells using marker genes
For the cells still assigned a Leiden cluster label, run a marker gene analysis
to determine the dominant cell type per cluster. We'll subset
larger clusters down to 10,000 cells to speed up the computation. Since scanpy's
`tl.rank_genes_groups` function needs normalizaed data, we'll use the log1p-transformed
matrix that we generated in @sec-preprocessing.
```{python}
#| label: L-4
#| echo: true
#| code-summary: "Python Code"
bdata = adata[adata.obs['leiden']==adata.obs['merged'], :].copy()
random_seed = 98103
np.random.seed(random_seed)
target_cells_per_cluster = 10000
cells_to_keep_indices = []
cluster_counts = bdata.obs['merged'].value_counts()
for cluster_label in cluster_counts.index:
cluster_cells = bdata.obs_names[bdata.obs['merged'] == cluster_label]
num_cells_in_cluster = len(cluster_cells)
if num_cells_in_cluster > target_cells_per_cluster:
print(f"Cluster '{cluster_label}' has {num_cells_in_cluster} cells, subsampling to {target_cells_per_cluster}.")
subsampled_cells = np.random.choice(
cluster_cells,
size=target_cells_per_cluster,
replace=False
)
cells_to_keep_indices.extend(subsampled_cells)
else:
cells_to_keep_indices.extend(cluster_cells)
bdata_subsampled = bdata[cells_to_keep_indices, :].copy()
bdata_subsampled.X = bdata_subsampled.layers['TC'].copy() # <1>
```
1. If you do not have the log1p-transformed total counts layer 'TC' saved, run `sc.pp.normalize_total`
and `sc.pp.log1p` (see @sec-tc-workflow).
Run `rank_genes_groups` and save results.
```{python}
#| label: run-marker-gene-leiden-2
#| echo: true
#| code-summary: "Python Code"
ct_dir = r.ct_dir
sc.settings.figdir = ct_dir
sc.tl.rank_genes_groups(bdata_subsampled, 'leiden', method='wilcoxon',
solver='saga', max_iter=200, pts=True) # ~an hour using logreg
sc.pl.rank_genes_groups(bdata_subsampled, n_genes=8, sharey=True, fontsize=10, save="_marker_gene_ranks_global.png")
sc.pl.rank_genes_groups_dotplot(bdata_subsampled, n_genes=8, save="_marker_gene_dotplot_global.png")
filename = os.path.join(r.analysis_dir, "anndata-5-subset-marker-genes.h5ad")
bdata_subsampled.write_h5ad(
filename,
compression=hdf5plugin.FILTERS["zstd"],
compression_opts=hdf5plugin.Zstd(clevel=5).filter_options
)
```
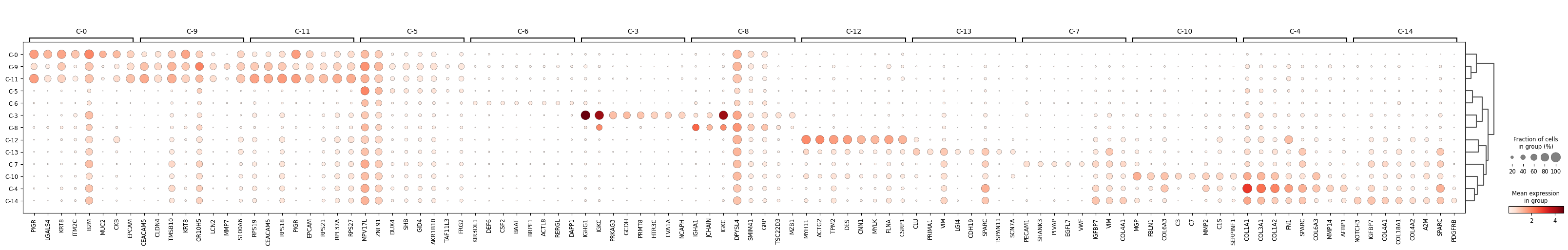
The next task is determine from the marker gene results the dominant cell types
that are likely found within each Leiden cluster. While some Leiden clusters
will have a high proportion of cells that are of the same type or state, keep
in mind that other Leiden clusters might be composites of cell types. The tools
we have for this exercise are:
- visual aids from the marker gene results and
- compare with external sources. The literature and subject matter expertise is the gold-standard for this.
```{r}
#| label: fig-global-marker-ranks
#| message: false
#| warning: false
#| echo: false
#| fig.width: 8
#| fig.height: 16
#| fig-cap: "Marker gene ranks by Leiden cluster (global)."
#| eval: true
render(file.path(analysis_asset_dir, "ct"), "rank_genes_groups_leiden_marker_gene_ranks_global.png", ct_dir, overwrite=TRUE)
```
```{r}
#| label: fig-global-marker-dot
#| message: false
#| warning: false
#| echo: false
#| fig.width: 12
#| fig.height: 5
#| fig-cap: "Marker gene dot plot by Leiden cluster (global)."
#| eval: true
render(file.path(analysis_asset_dir, "ct"), "dotplot__marker_gene_dotplot_global.png", ct_dir, overwrite = TRUE)
```
Examine the clusters by extracting the top 25 markers and their relative
scores and comparing to the literature. Large Language Models can provide supportive
information here but it's always recommended to use LLMs with caution and to verify
any of the results. In other words, LLMs can be fast but at the cost of accuracy.
In the example below, I created an LLM prompt that includes
the top marker gene statistics. Let's see what it generates.
```{python}
#| label: eval-marker-gene-leiden
#| echo: true
#| code-summary: "Python Code"
#| message: false
marker_dict, marker_df = format_ranked_genes(bdata_subsampled, 25)
csv_file_path = os.path.join(ct_dir, "colon_cancer_top_marker_genes_global.csv")
marker_df.to_csv(csv_file_path, index=True)
print(f"I am analyzing a CosMx SMI dataset of from Colon cancer and have run Leiden clustering.\nI have already classified several of the immune cells within the larger dataset and now I would like your help in predicting the most like cell type given the marker gene profile from this subset of data.\nBelow is a dictionary of results where the key represents the Leiden cluster\nname and its corresponding item is a list of ordered tuples where each tuple\nprovides the marker name and the relative score for that marker.\n\n Please provide a table with the columns that represent 1. inferred cell type 2. your confidence in that cell type 3. up to five markers from the list that are key to your result and 4. a brief justification of your result. Please present the results in a markdown table format to make it easier to copy and paste.\n\n{marker_dict}")
```
| Cluster | Inferred Cell Type | Confidence | Top 5 Key Markers | Brief Justification |
| :--- | :--- | :--- | :--- | :--- |
| **C-0** | **Epithelial (Goblet/Secretory)** | High | *PIGR, MUC2, EPCAM, KRT8, CLDN7* | Expresses broad epithelial markers (*EPCAM, KRT8*) alongside specific secretory/mucosal markers (*PIGR, MUC2*) typical of normal or well-differentiated colon epithelium. |
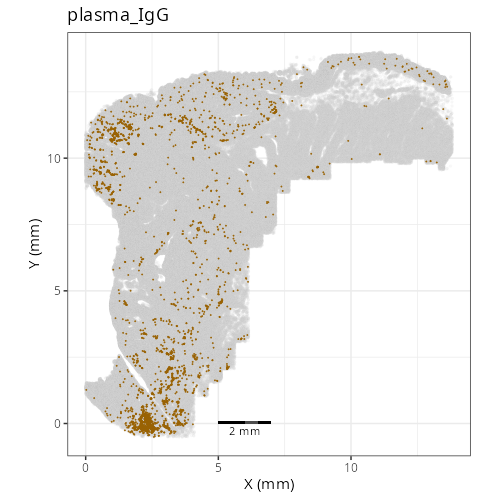
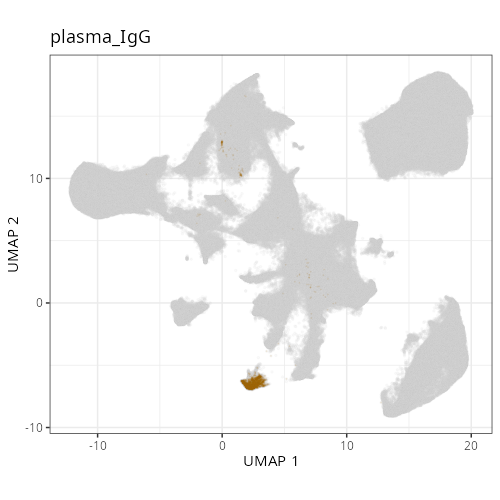
| **C-3** | **Plasma Cells (IgG)** | High | *IGHG1, IGKC, MZB1, JCHAIN, XBP1* | Defined by very high expression of Immunoglobulin G heavy chains (*IGHG1*) and plasma cell maturation factors (*MZB1, JCHAIN*). |
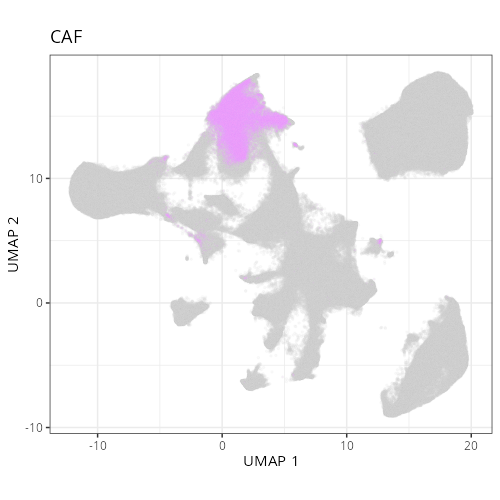
| **C-4** | **CAF** | High | *MMP14, MMP2, FN1, COL1A1, SPARC* | Spatially coincident with tumor. The high levels of matrix metalloproteinases (MMP14, MMP2) and Fibronectin (FN1) identify these as the activated myofibroblasts driving tumor desmoplasia. |
| **C-5** | **Unclassified** | Low | *MPV17L, ZNF91, DUX4, CXCL8, USP17L* | Lacks clear lineage markers (No *EPCAM, CD45,* or *COL1A1*). |
| **C-6** | **Immune (Mixed)** | Moderate | *KIR3DL1, CD79A, CSF2, CD53, RAG1* | Contains conflicting a mix of immune markers: NK cell (*KIR3DL1*) and B cell (*CD79A*) markers appear together. Likely represents lymphoid aggregates. |
| **C-7** | **Endothelial Cells** | High | *PECAM1, VWF, PLVAP, EGFL7, SHANK3* | Distinct expression of vascular endothelium markers, including *PECAM1* (CD31) and *VWF*. *PLVAP* suggests fenestrated capillaries common in the gut. |
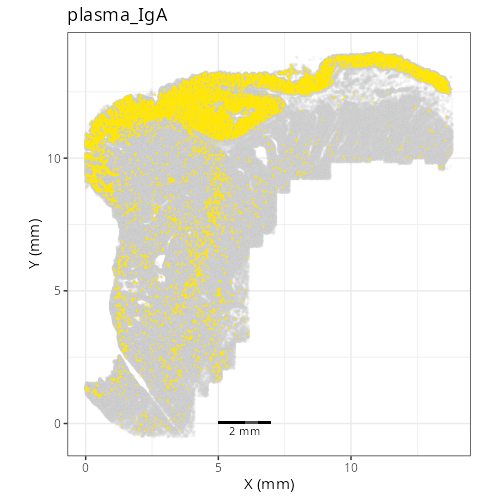
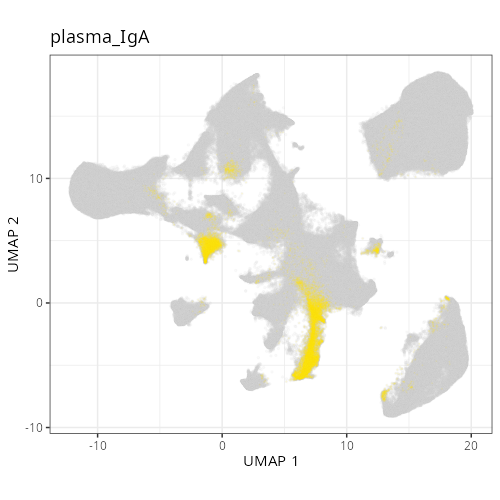
| **C-8** | **Plasma Cells (IgA)** | High | *IGHA1, JCHAIN, IGKC, GRP, MZB1* | Similar to C-3 but distinguished by the specific expression of IgA (*IGHA1*), the dominant isotype in mucosal immunity of the colon. |
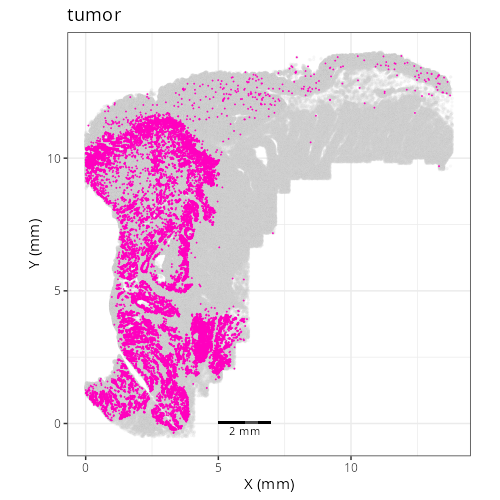
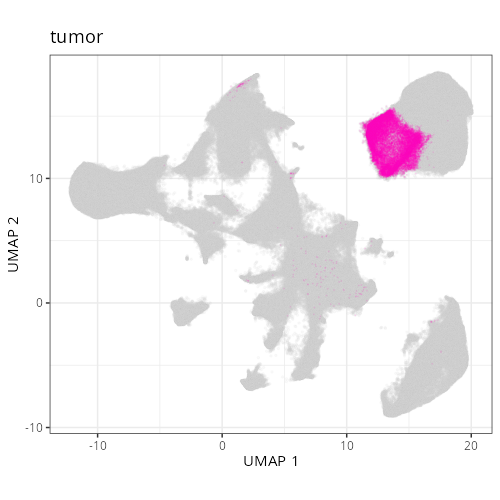
| **C-9** | **Tumor Epithelial (Invasive)** | High | *CEACAM5, MMP7, LCN2, KRT8, CLDN4* | Malignant epithelial profile (*EPCAM, KRT8*) with high expression of tumor markers (*CEACAM5/CEA*) and invasion/stress markers (*MMP7, LCN2*). |
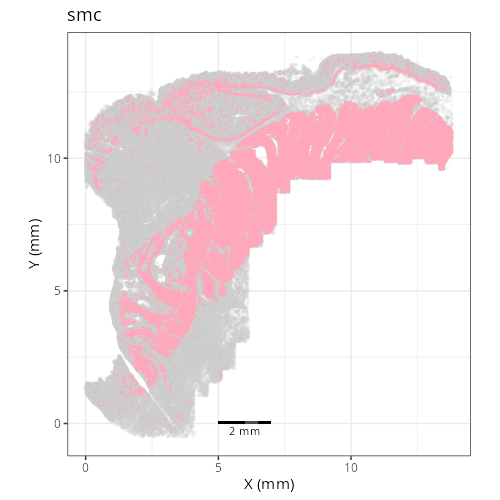
| **C-10** | **Structural Fibroblasts ** | High | *SFRP2, MGP, FBLN1, C3, CXCL12* | Spatially coincident with muscle (SMC). The profile (SFRP2+, MGP+, FBLN1+) matches "Adventitial" or "Universal" fibroblasts that act as the structural scaffold for muscle layers and vessels. |
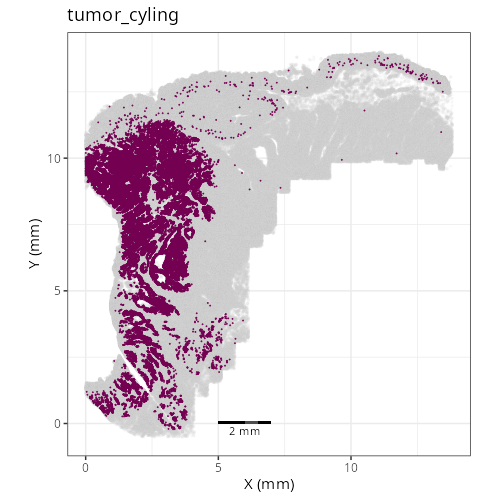
| **C-11** | **Tumor Epithelial (High Cycling)** | High | *RPS19, CEACAM5, EPCAM, RPS18, PIGR* | Epithelial tumor cells (*CEACAM5*) dominated by ribosomal proteins (*RPS/RPL*), indicating high protein synthesis and rapid cell cycling. |
| **C-12** | **Smooth Muscle Cells** | High | *MYH11, ACTG2, DES, CNN1, MYLK* | Expression of contractile proteins (*MYH11, ACTG2*) and Desmin (*DES*) identifies these as mural smooth muscle cells (muscularis mucosae/propria). |
| **C-13** | **Enteric Glia / Schwann Cells** | High | *CDH19, PMP22, SCN7A, LGI4, CLU* | Presence of peripheral nervous system markers (*PMP22, CDH19, SCN7A*) identifies these as supportive glial cells of the Enteric Nervous System. |
| **C-14** | **Pericytes** | High | *NOTCH3, PDGFRB, MCAM, COL4A1, RGS5* | Expresses pericyte markers (*PDGFRB, MCAM, NOTCH3*) and basement membrane collagen (*COL4A1*), distinguishing them from the broader fibroblast clusters. |
: Estimating most likely cell type from Leiden clusters' marker gene rankings. Note: predictions were generated with the aid of an LLM. {#tbl-leiden-global}
Following a little back and forth and helping provide the LLM with additional
spatial context, we have the marker gene results in @tbl-leiden-global, we have
most of the structural cell types
identified from the remaining clusters. Cluster C-5 lacks a clear and
dominant lineage signature. This cluster also occupies roughly the center of the UMAP
where poorer quality cells sometimes occupy. For the purposes of this tutorial
and the downstream questions, we'll leave those cells unclassified and label them
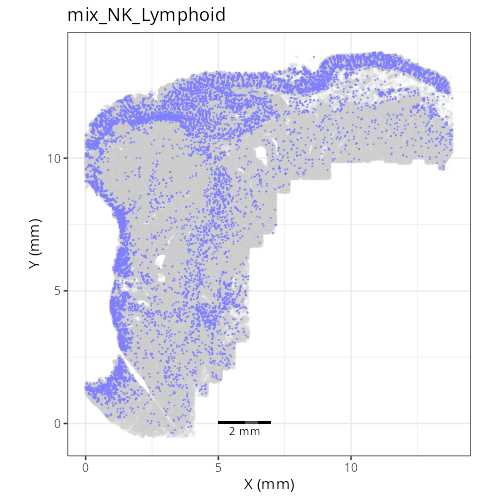
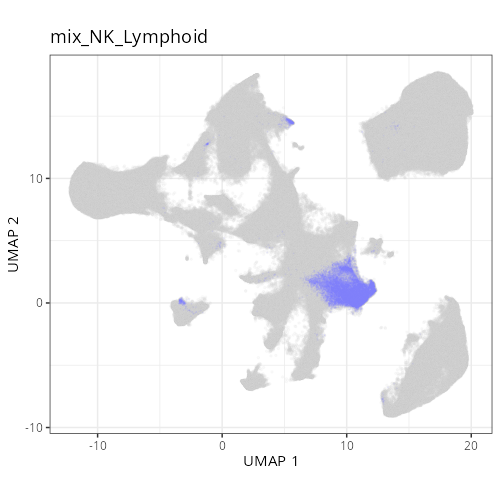
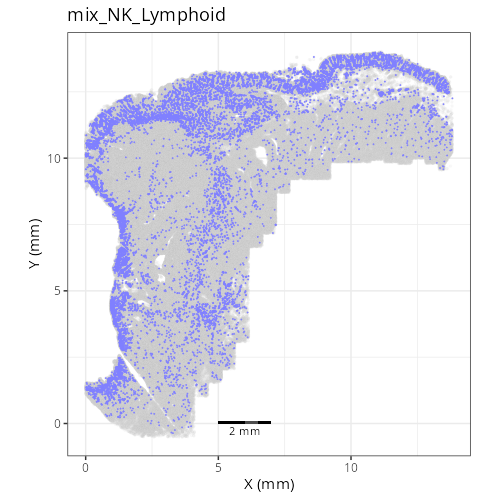
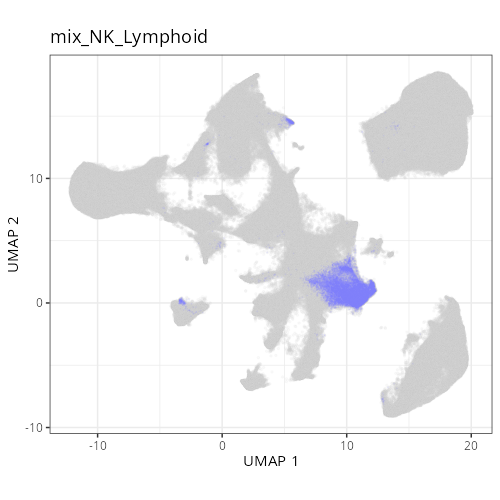
as "undetermined". Similarly, cluster C-6 showed two distinct lienage markers for
NK cells (_KIR3DL1_) and B cells (_CD79A_). Since these markers are unlikely to
be present in the same cell it is likely that C-6 comprises a composite of NK and
lymphoid cell types. Interestingly, HieraType did not classify the cells within
as either NK or B cell. Since our primary interest are on tumor and CAFs, we'll
not unravel this cluster any further and instead label it as a mix of cell types.
From the HieraType-derived merged data, map these new cell type labels.
```{python}
#| label: eval-marker-gene-leiden1
#| echo: true
#| code-summary: "Python Code"
#| message: false
#| eval: false
remap = {
'C-0': 'epithelial',
'C-3': 'plasma_IgG',
'C-4': 'CAF',
'C-5': 'undetermined',
'C-6': 'mix_NK_Lymphoid',
'C-7': 'endothelial',
'C-8': 'plasma_IgA',
'C-9': 'tumor',
'C-10': 'fibroblast',
'C-11': 'tumor_cyling',
'C-12': 'smc',
'C-13': 'glial',
'C-14': 'pericyte'
}
adata.obs['celltype_fine'] = adata.obs['merged'].astype(str).replace(remap)
```
Often we want to plot or work with broader classifications of cell types. The code
below combines, for example, all the various T cell subtypes into a single group.
```{python}
#| label: H-6
#| eval: false
#| code-summary: "Python Code"
#| message: true
#| warning: false
#| code-fold: show
broad_map = {
'cd8_tem': 'tcell',
'cd4_treg': 'tcell',
'cd4_tem': 'tcell',
'cd8_cytotoxic': 'tcell',
'cd8_naive': 'tcell',
'cd8_exhausted': 'tcell',
'cd4_tcm': 'tcell',
'cd4_th2': 'tcell',
'cd4_th17': 'tcell',
'cd4_naive': 'tcell',
'cd4_th1': 'tcell',
'cd8_tcm': 'tcell',
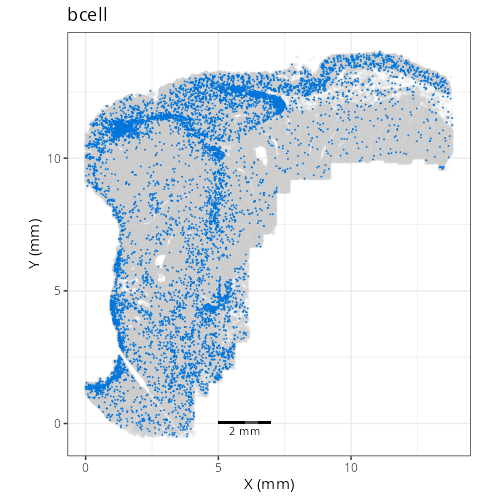
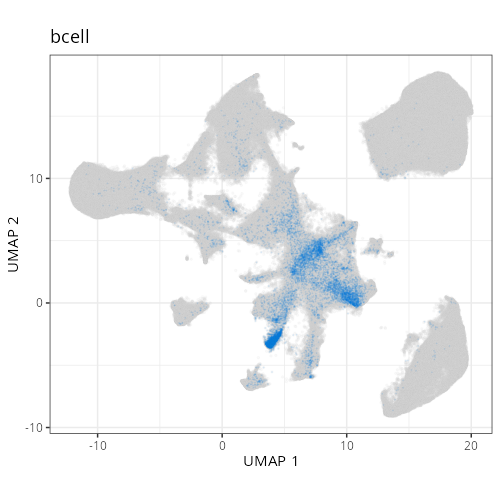
'bcell': 'bcell',
'plasma_IgA': 'plasma',
'plasma_IgG': 'plasma',
'tumor': 'tumor',
'tumor_cyling': 'tumor',
'epithelial': 'epithelial',
'CAF': 'CAF',
'fibroblast': 'fibroblast',
'smc': 'smc',
'pericyte': 'pericyte',
'endothelial': 'endothelial',
'glial': 'glial',
'macrophage': 'macrophage',
'monocyte': 'monocyte',
'dendritic': 'dendritic',
'mast': 'mast',
'neutrophil': 'neutrophil',
'nk': 'nk',
'mix_NK_Lymphoid': 'mix_NK_Lymphoid',
'undetermined': 'undetermined'
}
adata.obs['celltype_broad'] = adata.obs['celltype_fine'].map(broad_map)
```
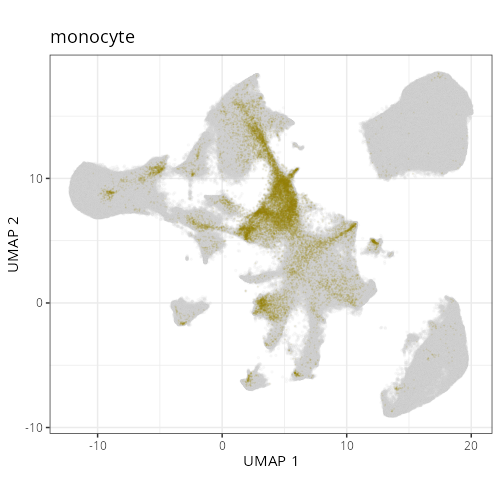
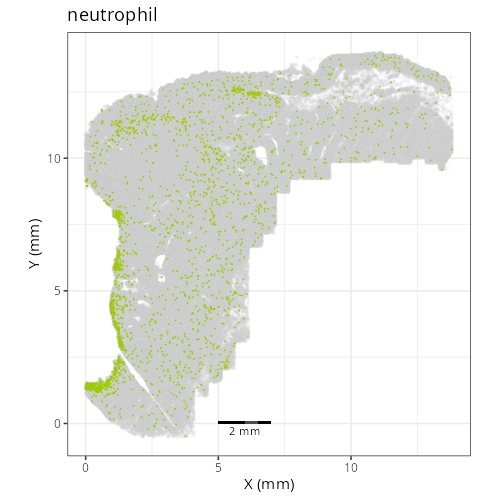
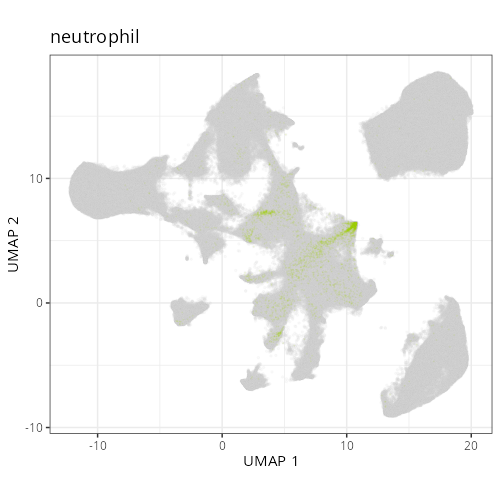
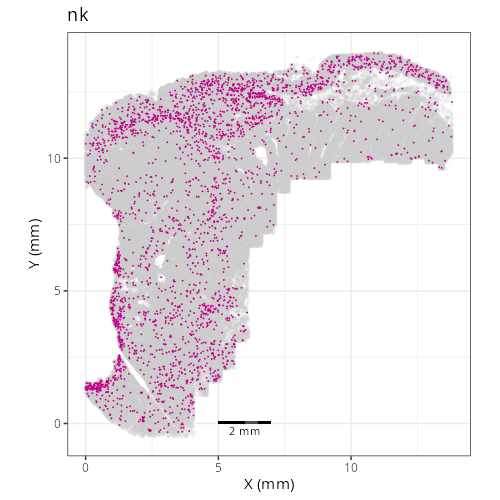
Which provides 18 broad cell types and 32 fine cell types.
Save the results to disk.
```{python}
#| label: H-7
#| eval: false
#| code-summary: "R Code"
#| message: true
#| warning: false
#| code-fold: show
filename = os.path.join(r.analysis_dir, "anndata-6-celltypes.h5ad")
adata.write_h5ad(
filename,
compression=hdf5plugin.FILTERS["zstd"],
compression_opts=hdf5plugin.Zstd(clevel=5).filter_options
)
```
## Cell Type Visualization
It is useful at this point to visualize the distributions of cells in a variety
of ways to get an understanding of their composition. We can visualize these
cell types in UMAP space and XY space.
### Broad cell types
Instead of the default colors in `plotDots`, let's create manual ones.
```{r}
#| label: V-1
#| eval: false
#| code-summary: "R Code"
#| code-fold: true
# define colors semi-manually instead of using the default colors
column <- 'celltype_broad'
column_levels <- levels(py$adata$obs[[column]])
n_levels <- length(column_levels)
if(n_levels<27){
colors_use <- pals::alphabet(n_levels)
} else if(n < 53){
colors_use <- c(pals::alphabet(26), pals::alphabet2(n_levels-26))
} else {
stop("consider fewer groups")
}
names(colors_use) <- column_levels
colors_use['mast'] = "#FFCC99"
colors_use['tumor'] = "#C20088"
colors_use['undetermined'] = "#808080"
colors_use['mix_NK_Lymphoid'] = "#8080FF"
# saving colors for later
results_list[['celltype_broad_names']] <- names(colors_use)
results_list[['celltype_broad_colors']] <- as.character(colors_use)
saveRDS(results_list, results_list_file)
```
Plot the cells in XY space and UMAP space and color based on `celltype_broad`.
```{r}
#| label: V-2
#| eval: false
#| code-summary: "R Code"
#| code-fold: true
group_prefix <- "broad" # <1>
ct_assets_dir <- file.path(analysis_asset_dir, "ct")
# XY (global only)
plotDots(py$adata, color_by='celltype_broad',
plot_global = TRUE,
facet_by_group = FALSE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001
),
scale_bar_params = list(
location = c(5, 0),
width = 2,
n = 3,
height = 0.1,
scale_colors = c("black", "grey30"),
label_nudge_y = -0.3
),
directory = ct_assets_dir,
fileType = "png",
dpi = 200,
width = 8,
height = 8,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("X (mm)"),
ylab("Y (mm)"),
coord_fixed(),
scale_color_manual(values = colors_use),
theme(legend.position = c(0.8, 0.4)),
guides(color = guide_legend(
title="Cell Type",
override.aes = list(size = 3) ) )
)
)
# XY (facets only)
plotDots(py$adata, color_by='celltype_broad',
plot_global = FALSE,
facet_by_group = TRUE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001
),
scale_bar_params = list(
location = c(5, 0),
width = 2,
n = 3,
height = 0.1,
scale_colors = c("black", "grey30"),
label_nudge_y = -0.3
),
directory = ct_assets_dir,
fileType = "png",
dpi = 100,
width = 5,
height = 5,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("X (mm)"),
ylab("Y (mm)"),
coord_fixed(),
scale_color_manual(values = colors_use),
theme(legend.position = c(0.8, 0.4)),
guides(color = guide_legend(
title="Cell Type",
override.aes = list(size = 3) ) )
)
)
# UMAP (global only)
plotDots(py$adata,
obsm_key = "umap_pr",
color_by='celltype_broad',
plot_global = TRUE,
facet_by_group = FALSE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001, alpha=0.1
),
geom_label_params = list(
size = 2
),
labels_on_plot = data.frame(),
directory = ct_assets_dir,
fileType = "png",
dpi = 200,
width = 8,
height = 8,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("UMAP 1"),
ylab("UMAP 2"),
coord_fixed(),
scale_color_manual(values = colors_use),
# guides(color = guide_legend(
# title="Cell Type",
# override.aes = list(size = 3) ) ),
theme(legend.position = "none")
)
)
# UMAP (facets only)
plotDots(py$adata,
obsm_key = "umap_pr",
color_by='celltype_broad',
plot_global = FALSE,
facet_by_group = TRUE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001, alpha=0.1
),
geom_label_params = list(
size = 2
),
labels_on_plot = data.frame(),
directory = ct_assets_dir,
fileType = "png",
dpi = 100,
width = 5,
height = 5,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("UMAP 1"),
ylab("UMAP 2"),
coord_fixed(),
scale_color_manual(values = colors_use),
guides(color = guide_legend(
title="Cell Type",
override.aes = list(size = 3) ) )
)
)
```
1. in the plotting below, we will use this name to parse out files.
Here are the overall (global) plots.
::: {.panel-tabset}
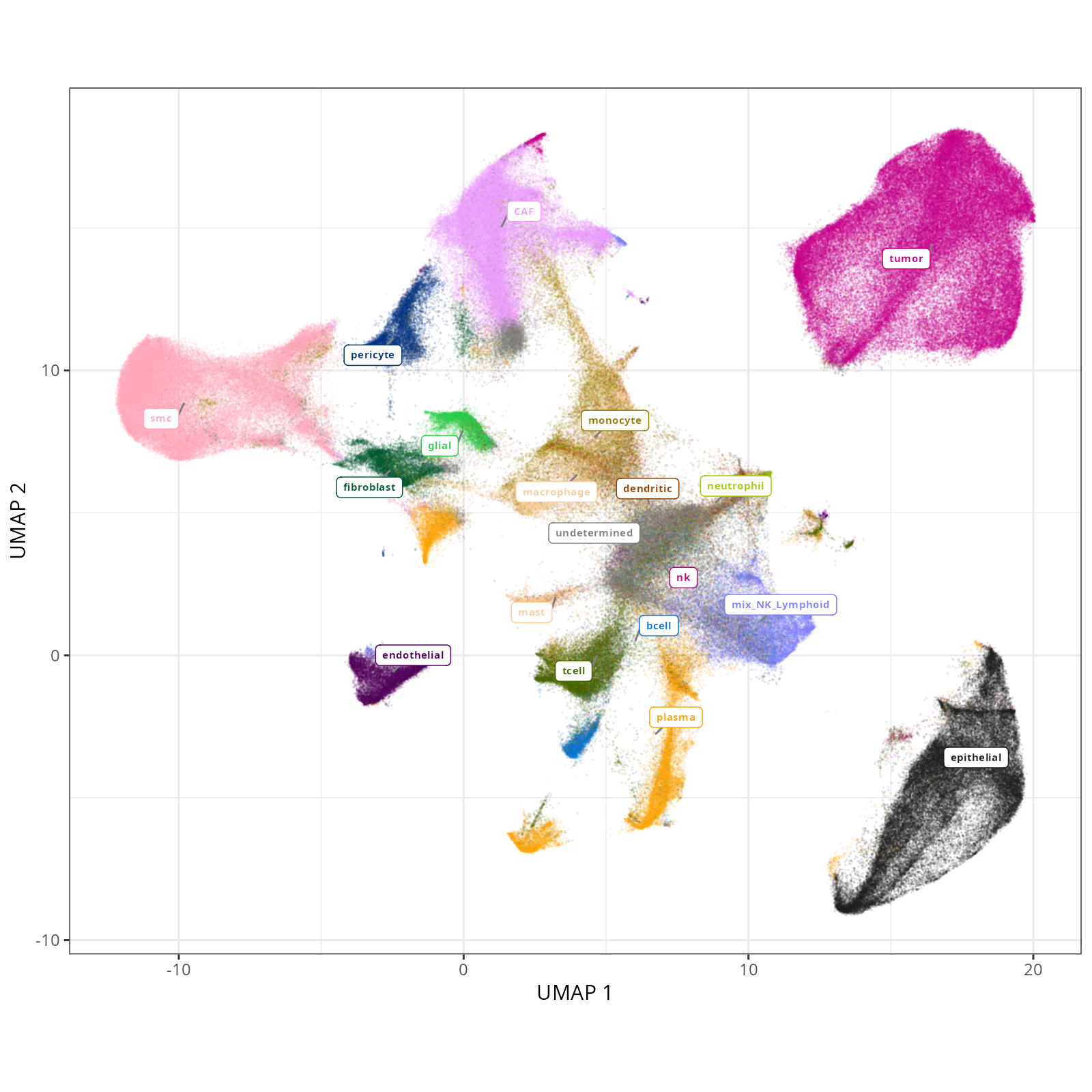
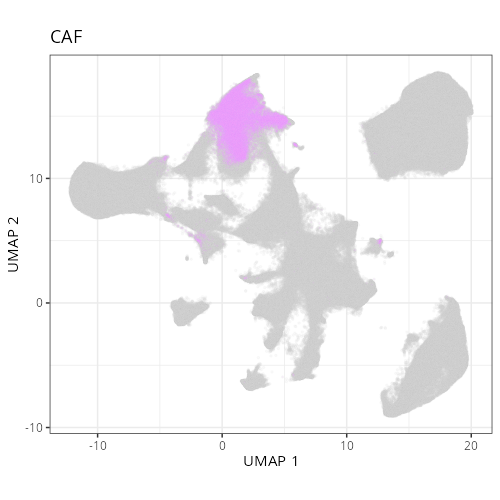
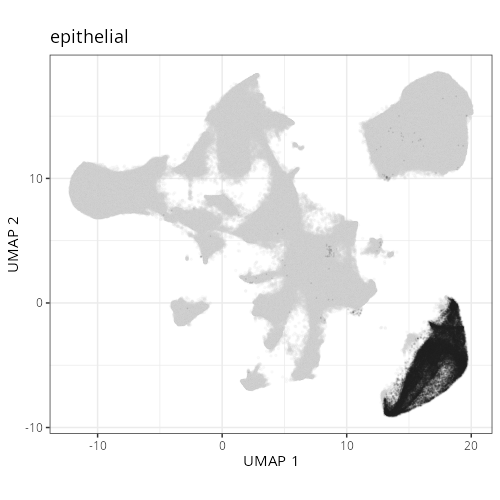
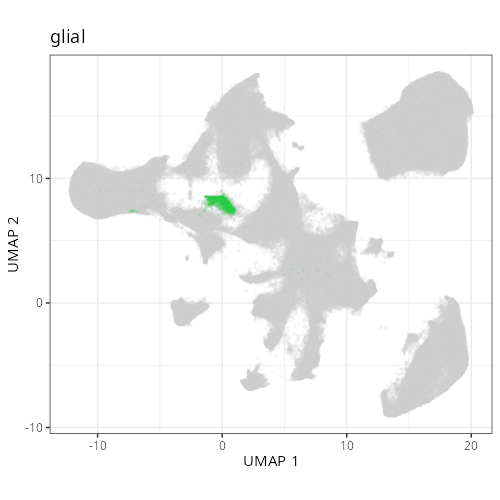
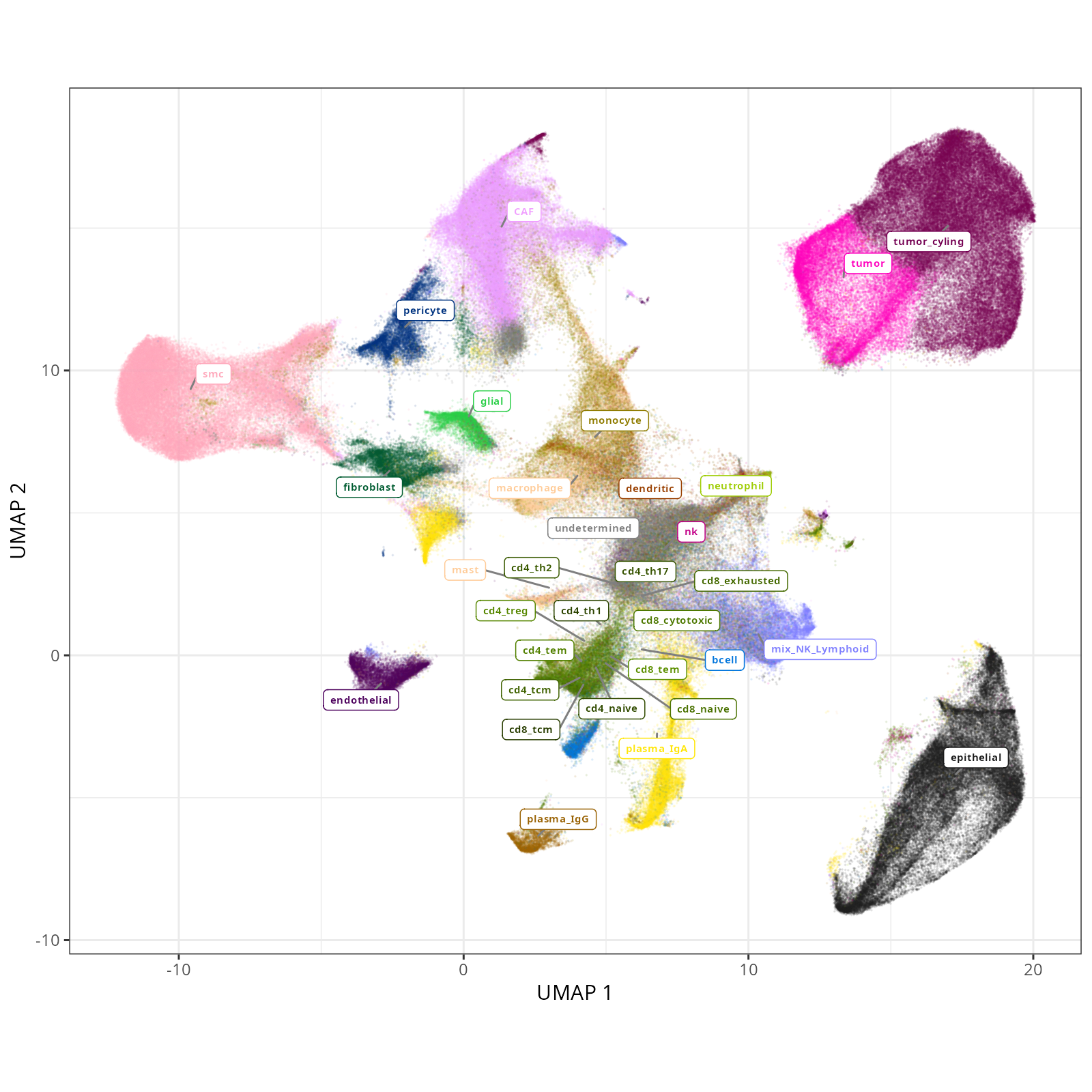
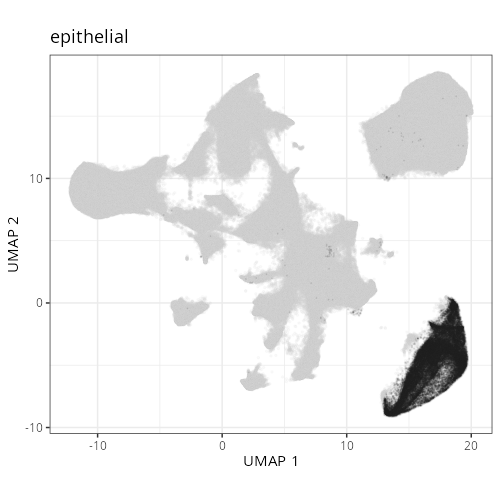
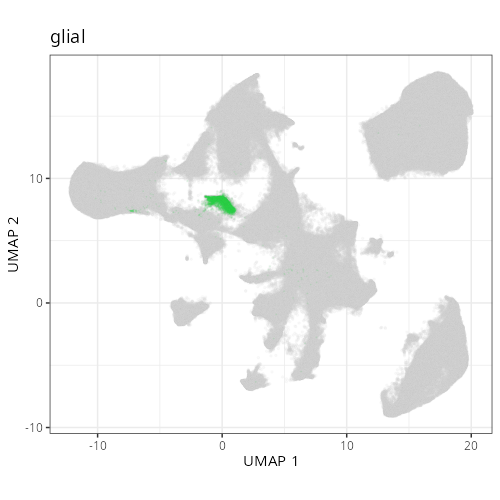
#### Cell Type Broad - UMAP
```{r}
#| label: fig-umap-celltype-broad
#| message: false
#| warning: false
#| echo: false
#| fig.width: 6
#| fig.height: 5
#| fig-cap: "UMAP with broad cell type in the full dataset."
#| eval: true
knitr::include_graphics(file.path(analysis_asset_dir, "ct", "broad__umap_pr__S0__plot.png"))
```
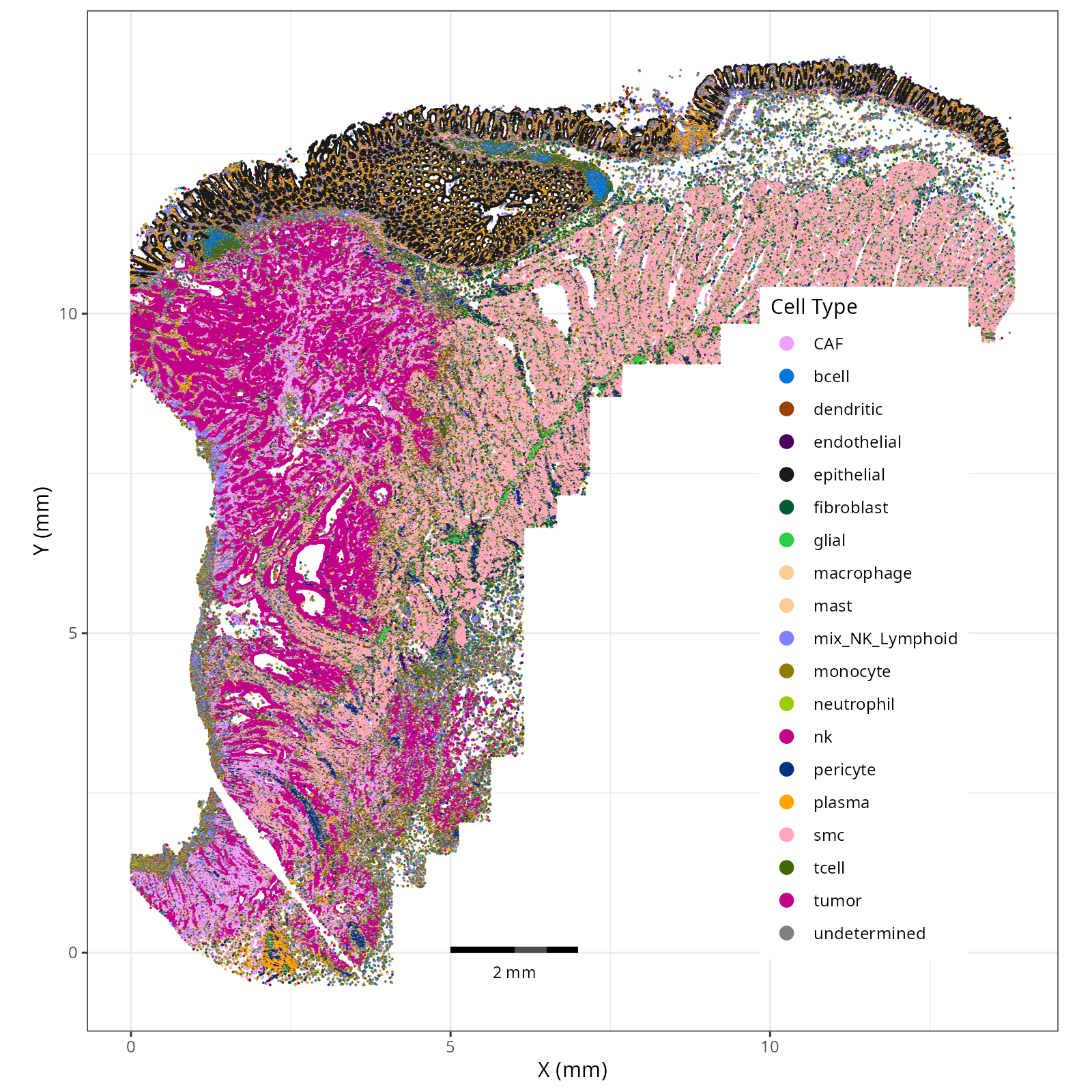
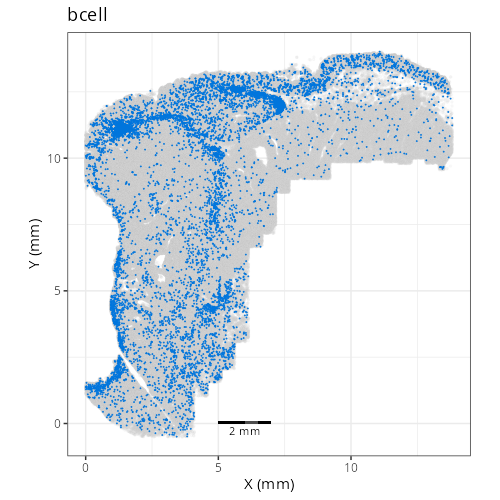
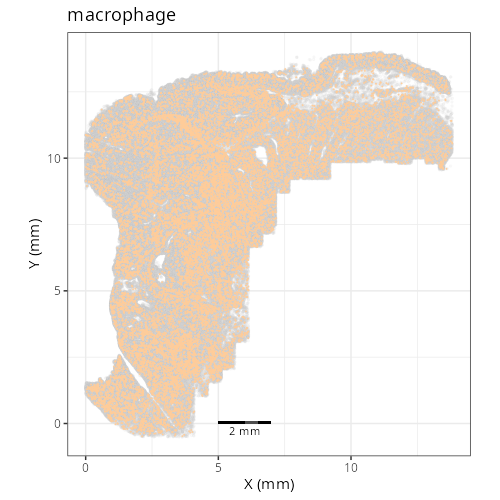
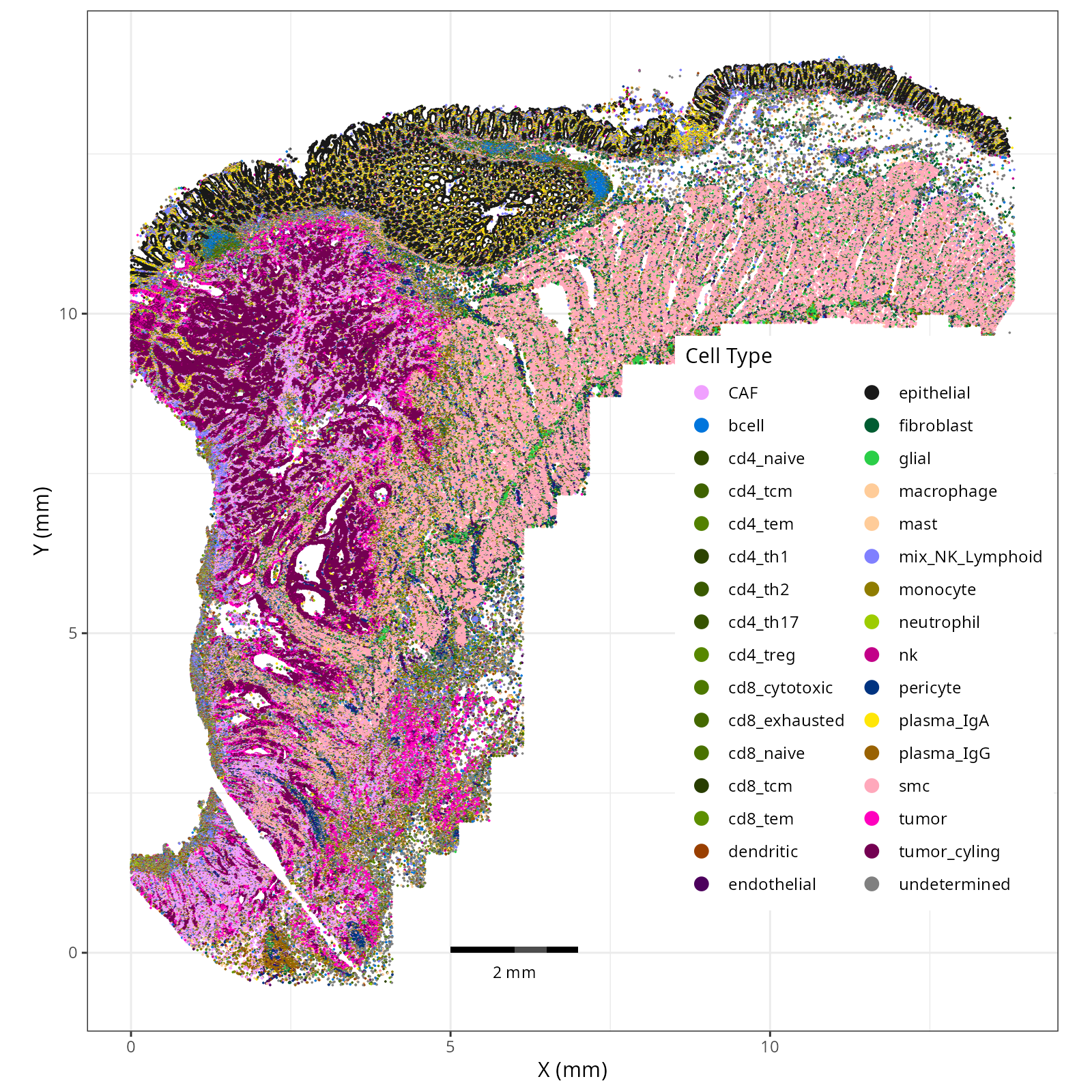
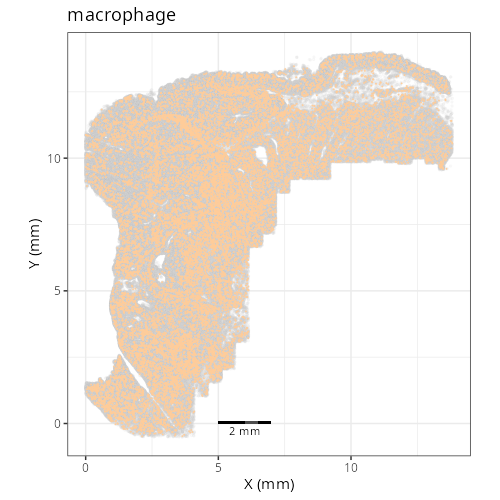
#### Cell Type Broad - XY
```{r}
#| label: fig-xy-celltype-broad
#| message: false
#| warning: false
#| echo: false
#| fig.width: 6
#| fig.height: 5
#| fig-cap: "XY with broad cell type in the full dataset."
#| eval: true
knitr::include_graphics(file.path(analysis_asset_dir, "ct", "broad__spatial__S0__plot.png"))
```
:::
And here are the facetted plots.
::: {.panel-tabset}
```{r}
#| output: asis
#| eval: true
#| code-fold: true
#| echo: true
fig_dir <- file.path(analysis_asset_dir, "ct")
group_prefix <- "broad"
slide_id <- "S0"
xy_files <- Sys.glob(file.path(fig_dir,
paste0(group_prefix,
"__spatial__", slide_id, "__facet_*png")))
umap_files <- Sys.glob(file.path(fig_dir,
paste0(group_prefix,
"__umap_pr__", slide_id, "__facet_*png")))
res <- purrr::map2_chr(xy_files, umap_files, \(current_xy_file, current_umap_file){
knitr::knit_child(text = c(
"### `r gsub('.png', '', strsplit(current_umap_file, split='__facet_')[[1]][2])`",
"",
":::{columns}",
"",
"::: {.column width='40%'}",
"",
"```{r eval=TRUE}",
"#| echo: false",
"knitr::include_graphics(current_xy_file)",
"```",
"",
":::",
"",
"::: {.column width='10%'}",
"",
":::",
"",
"::: {.column width='40%'}",
"",
"```{r eval=TRUE}",
"#| echo: false",
"knitr::include_graphics(current_umap_file)",
"```",
":::",
"",
":::",
"",
"",
""
), envir = environment(), quiet = TRUE)
})
cat(res, sep = '\n')
```
:::
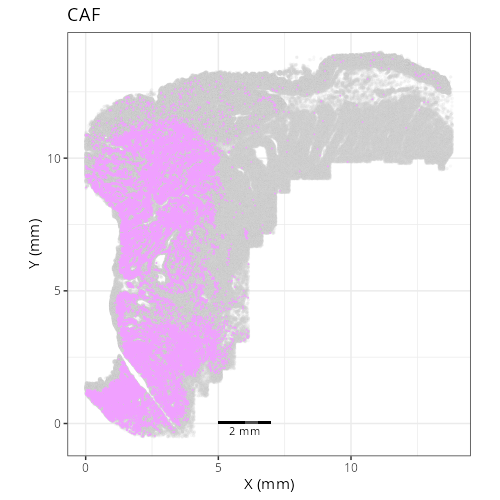
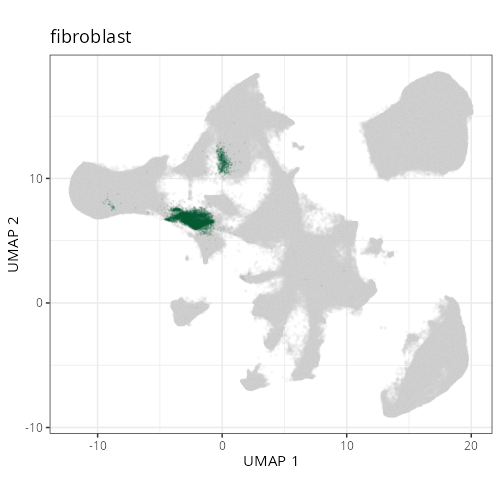
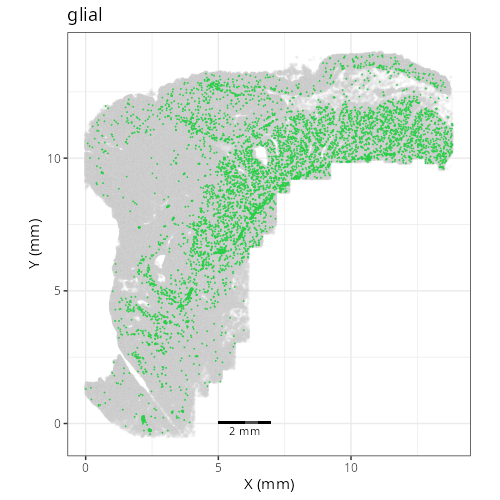
### Fine cell types
This section performs a similar routine as above but with the fine cell type
classifications.
For the fine cell types, we'll create colors that are various shades of the
parent type.
```{r}
#| label: V-3
#| eval: false
#| code-summary: "R Code"
#| code-fold: true
broad_map <- unlist(py$broad_map)
generate_fine_colors <- function(mapping, broad_colors, mode = "shades") {
groups <- split(names(mapping), mapping)
fine_colors <- c()
for (broad_name in names(groups)) {
subtypes <- groups[[broad_name]]
n <- length(subtypes)
base_col <- broad_colors[broad_name]
if (n == 1) {
cols <- base_col
names(cols) <- subtypes
} else {
if (mode == "shades") {
ramp_func <- colorRampPalette(c(
adjustcolor(base_col, transform=diag(c(1,1,1,1))*1.4), # Lighten
base_col,
adjustcolor(base_col, transform=diag(c(1,1,1,1))*0.6) # Darken
))
cols <- ramp_func(n)
} else {
ramp_func <- colorRampPalette(c("#FFFFFF", base_col, "#000000"))
cols <- ramp_func(n + 2)[2:(n + 1)]
}
names(cols) <- subtypes
}
fine_colors <- c(fine_colors, cols)
}
return(fine_colors)
}
fine_colors <- generate_fine_colors(broad_map, colors_use)
results_list[['celltype_fine_names']] <- names(fine_colors)
results_list[['celltype_fine_colors']] <- as.character(fine_colors)
saveRDS(results_list, results_list_file)
```
```{r}
#| eval: false
#| code-summary: "R Code"
#| code-fold: true
group_prefix <- "fine" # <1>
ct_assets_dir <- file.path(analysis_asset_dir, "ct")
# XY (global only)
plotDots(py$adata, color_by='celltype_fine',
plot_global = TRUE,
facet_by_group = FALSE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001
),
scale_bar_params = list(
location = c(5, 0),
width = 2,
n = 3,
height = 0.1,
scale_colors = c("black", "grey30"),
label_nudge_y = -0.3
),
directory = ct_assets_dir,
fileType = "png",
dpi = 200,
width = 8,
height = 8,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("X (mm)"),
ylab("Y (mm)"),
coord_fixed(),
scale_color_manual(values = fine_colors),
theme(legend.position = c(0.8, 0.4)),
guides(color = guide_legend(
title="Cell Type",
override.aes = list(size = 3) ) )
)
)
# XY (facets only)
plotDots(py$adata, color_by='celltype_fine',
plot_global = FALSE,
facet_by_group = TRUE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001
),
scale_bar_params = list(
location = c(5, 0),
width = 2,
n = 3,
height = 0.1,
scale_colors = c("black", "grey30"),
label_nudge_y = -0.3
),
directory = ct_assets_dir,
fileType = "png",
dpi = 100,
width = 5,
height = 5,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("X (mm)"),
ylab("Y (mm)"),
coord_fixed(),
scale_color_manual(values = fine_colors),
theme(legend.position = c(0.8, 0.4)),
guides(color = guide_legend(
title="Cell Type",
override.aes = list(size = 3) ) )
)
)
# UMAP (global only)
plotDots(py$adata,
obsm_key = "umap_pr",
color_by='celltype_fine',
plot_global = TRUE,
facet_by_group = FALSE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001, alpha=0.1
),
geom_label_params = list(
size = 2
),
labels_on_plot = data.frame(),
directory = ct_assets_dir,
fileType = "png",
dpi = 200,
width = 8,
height = 8,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("UMAP 1"),
ylab("UMAP 2"),
coord_fixed(),
scale_color_manual(values = fine_colors),
# guides(color = guide_legend(
# title="Cell Type",
# override.aes = list(size = 3) ) ),
theme(legend.position = "none")
)
)
# UMAP (facets only)
plotDots(py$adata,
obsm_key = "umap_pr",
color_by='celltype_fine',
plot_global = FALSE,
facet_by_group = TRUE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001, alpha=0.1
),
geom_label_params = list(
size = 2
),
labels_on_plot = data.frame(),
directory = ct_assets_dir,
fileType = "png",
dpi = 100,
width = 5,
height = 5,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("UMAP 1"),
ylab("UMAP 2"),
coord_fixed(),
scale_color_manual(values = fine_colors),
guides(color = guide_legend(
title="Cell Type",
override.aes = list(size = 3) ) )
)
)
```
::: {.panel-tabset}
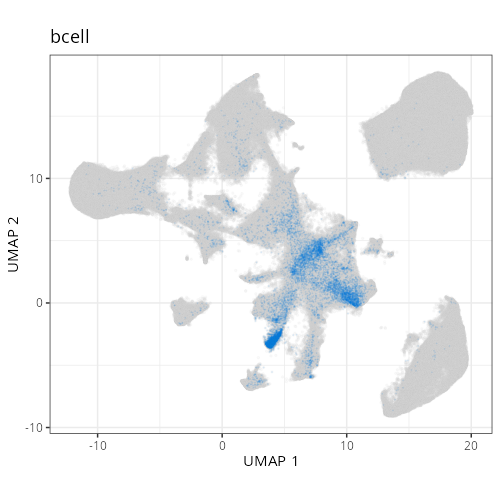
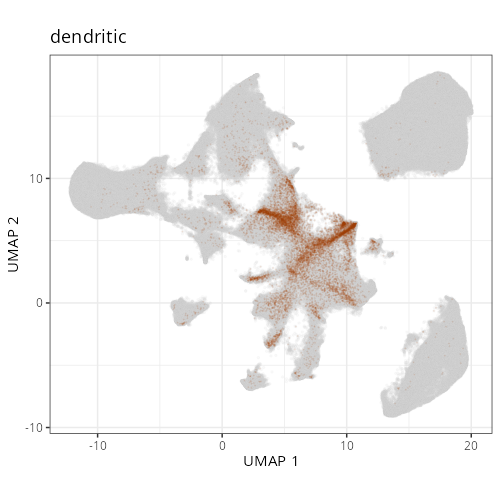
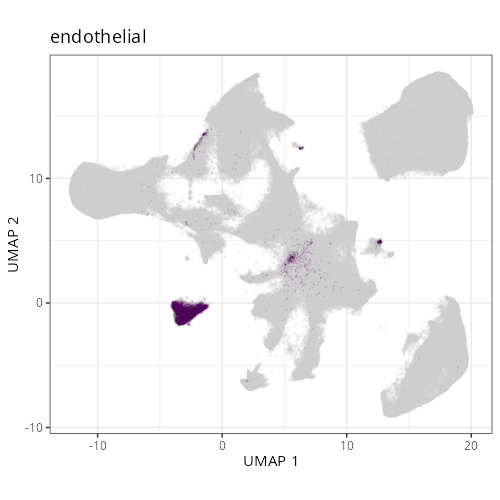
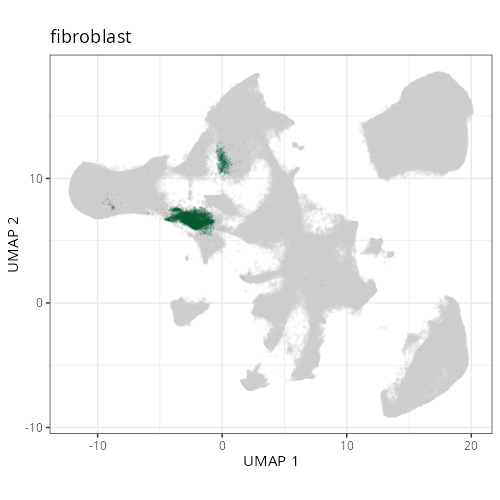
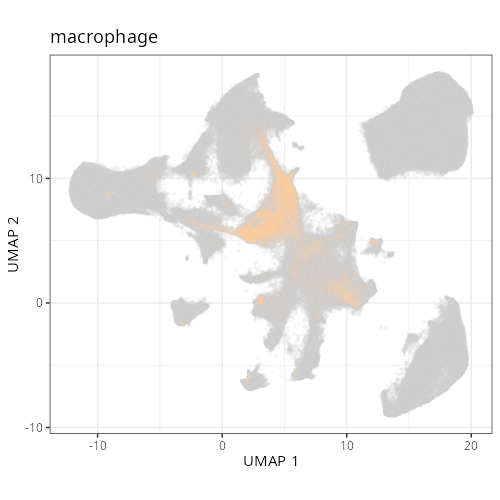
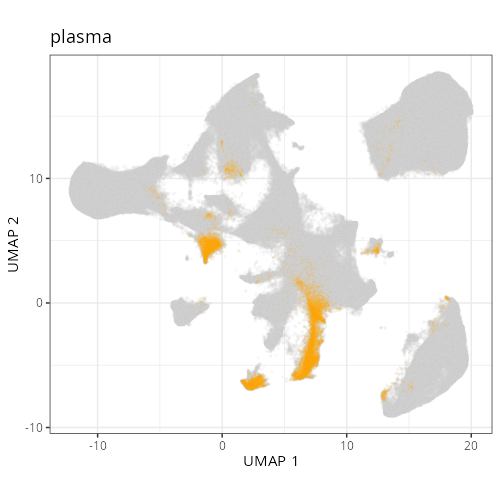
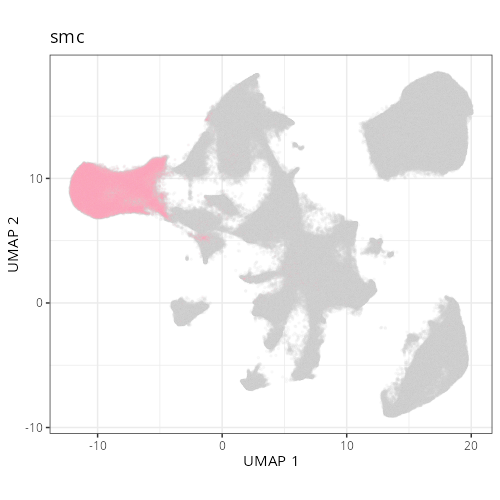
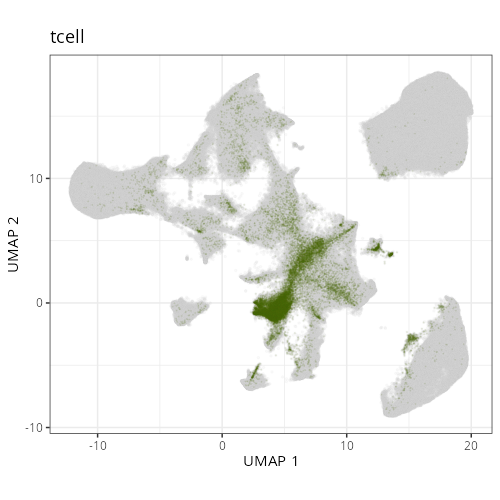
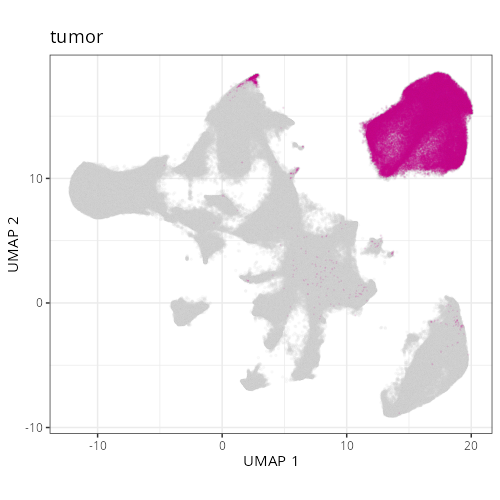
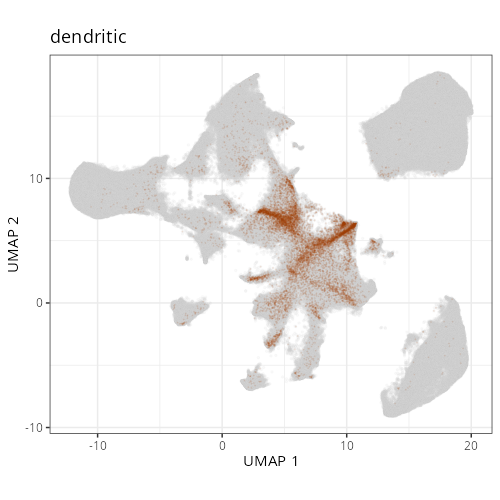
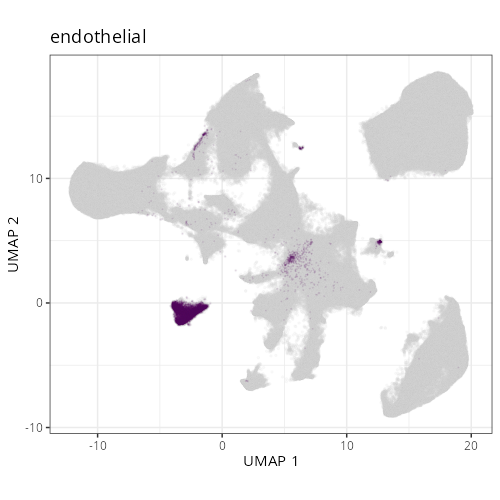
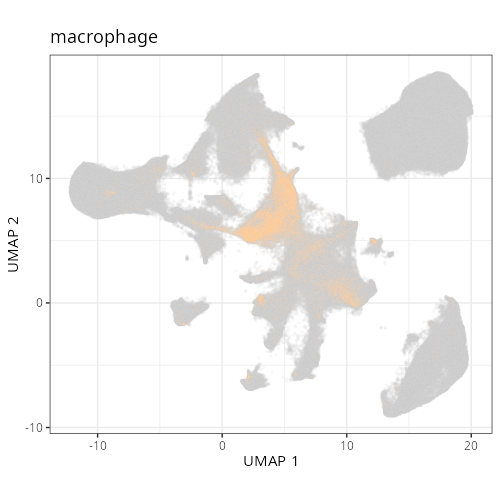
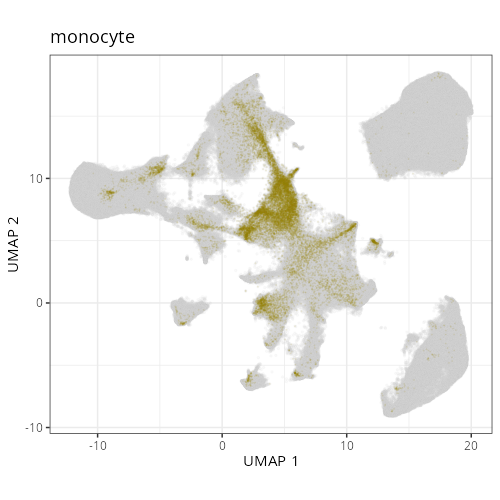
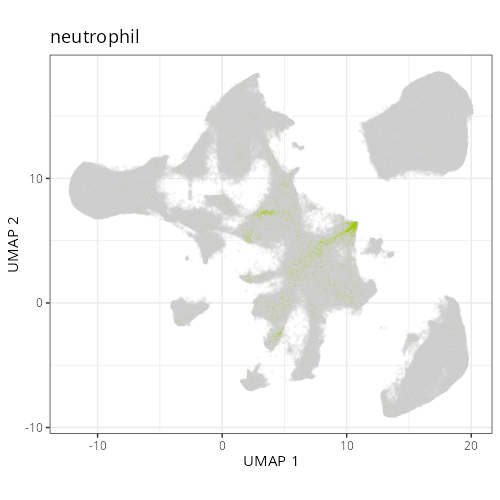
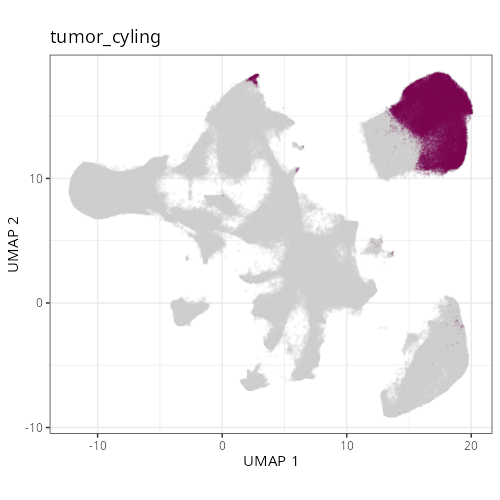
#### Cell Type Fine - UMAP
```{r}
#| label: fig-umap-celltype-fine
#| message: false
#| warning: false
#| echo: false
#| fig.width: 6
#| fig.height: 5
#| fig-cap: "UMAP with fine cell type labels."
#| eval: true
knitr::include_graphics(file.path(analysis_asset_dir, "ct", "fine__umap_pr__S0__plot.png"))
```
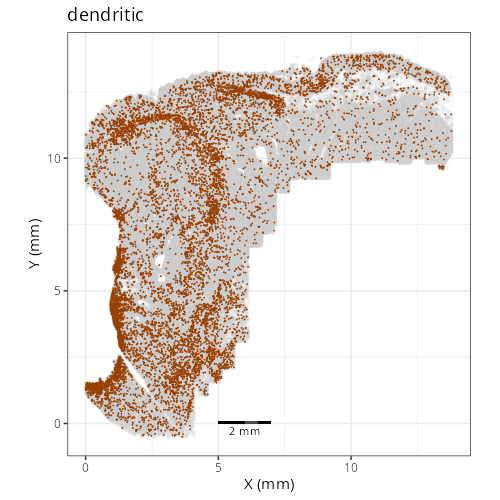
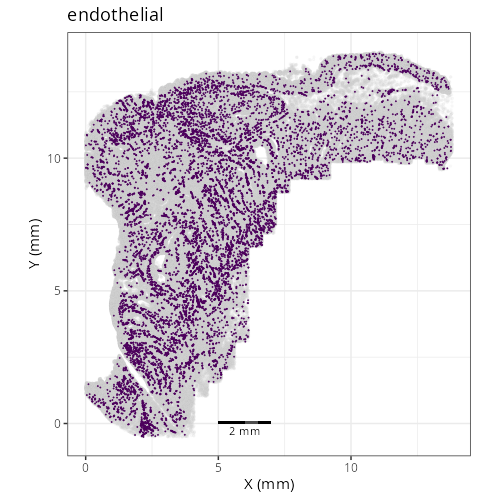
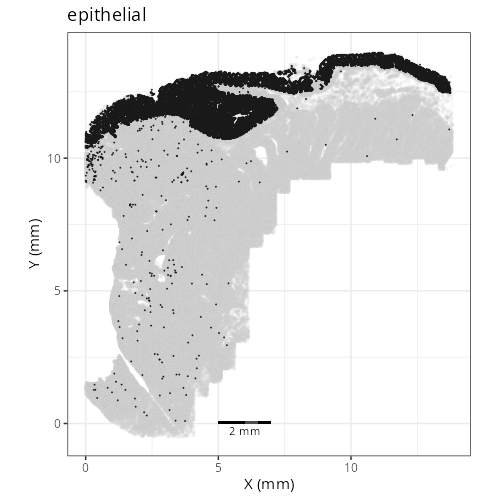
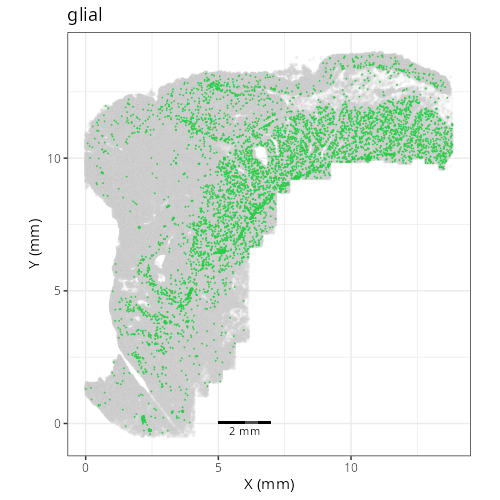
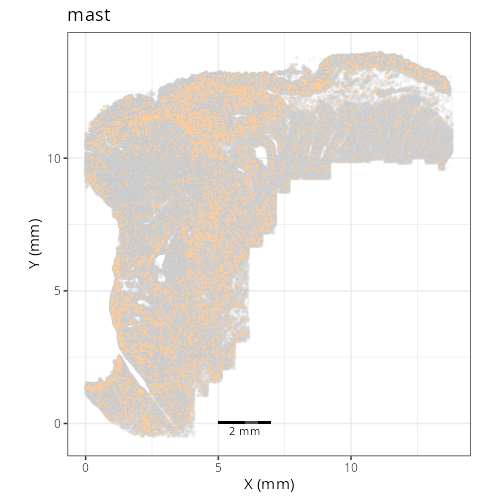
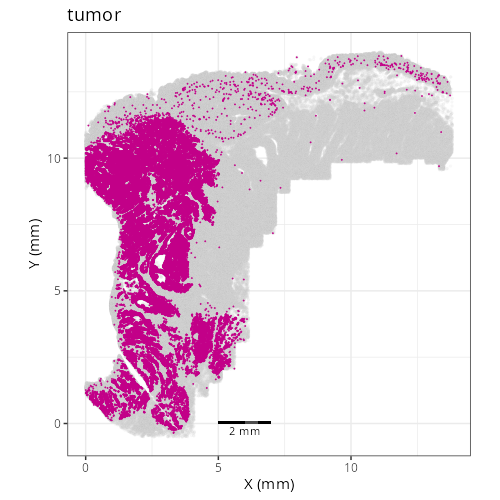
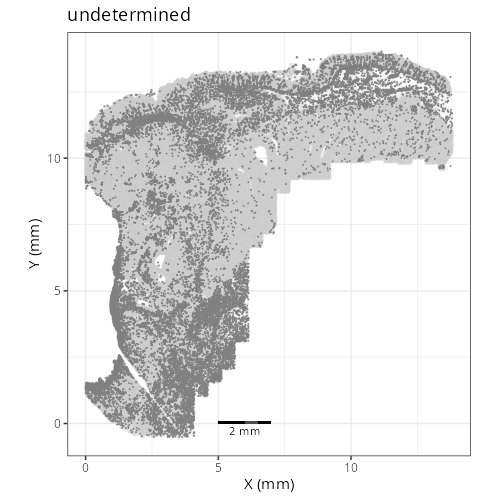
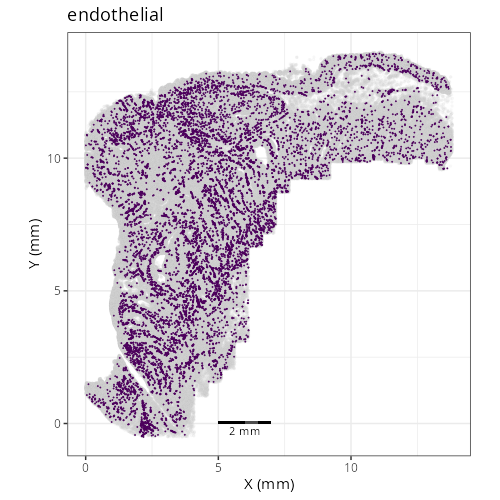
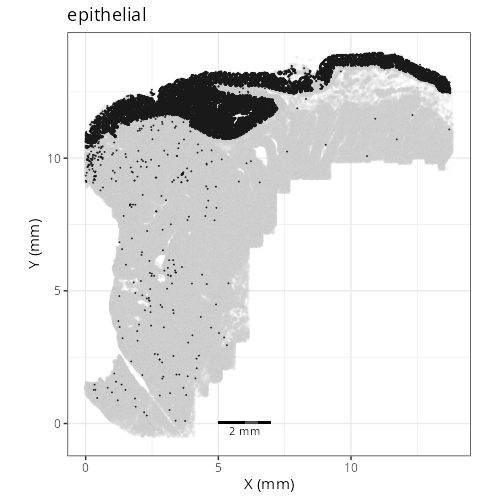
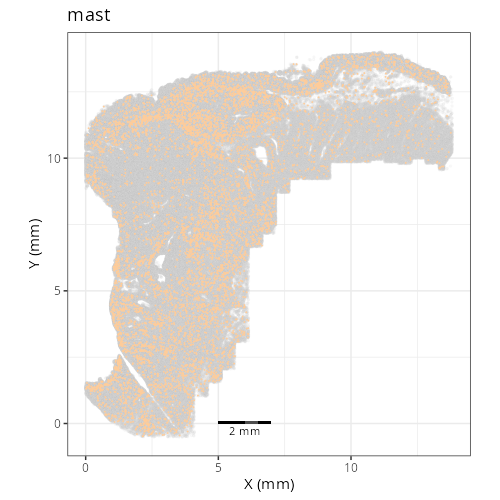
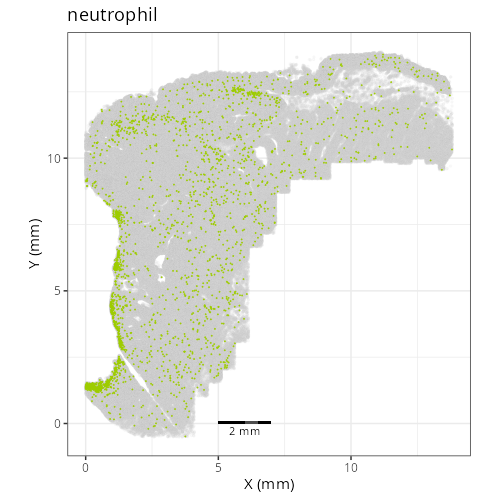
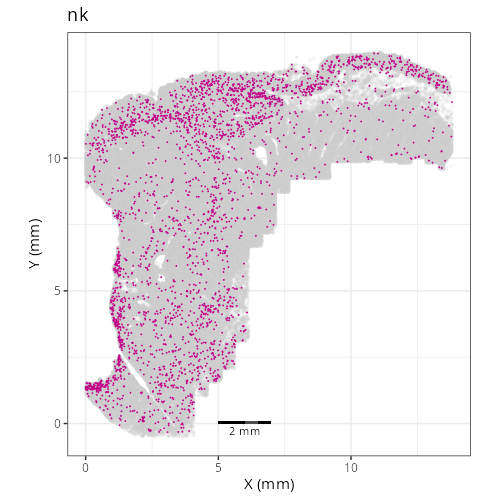
#### Cell Type Fine - XY
```{r}
#| label: fig-xy-celltype-fine
#| message: false
#| warning: false
#| echo: false
#| fig.width: 6
#| fig.height: 5
#| fig-cap: "XY with fine cell type labels."
#| eval: true
knitr::include_graphics(file.path(analysis_asset_dir, "ct", "fine__spatial__S0__plot.png"))
```
:::
And here are the facetted plots.
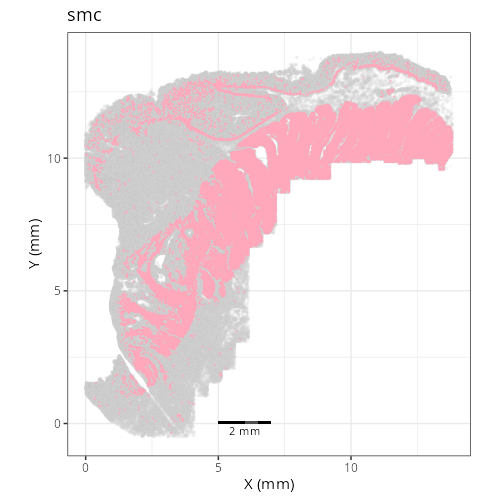
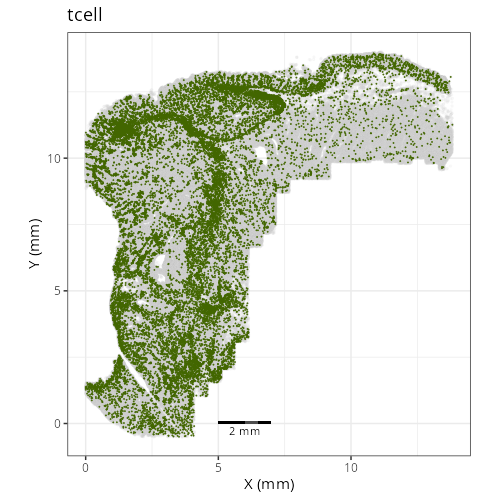
::: {.panel-tabset}
```{r}
#| output: asis
#| eval: true
#| code-fold: true
#| echo: true
fig_dir <- file.path(analysis_asset_dir, "ct")
group_prefix <- "fine"
slide_id <- "S0"
xy_files <- Sys.glob(file.path(fig_dir,
paste0(group_prefix,
"__spatial__", slide_id, "__facet_*png")))
umap_files <- Sys.glob(file.path(fig_dir,
paste0(group_prefix,
"__umap_pr__", slide_id, "__facet_*png")))
res <- purrr::map2_chr(xy_files, umap_files, \(current_xy_file, current_umap_file){
knitr::knit_child(text = c(
"### `r gsub('.png', '', strsplit(current_umap_file, split='__facet_')[[1]][2])`",
"",
":::{columns}",
"",
"::: {.column width='40%'}",
"",
"```{r eval=TRUE}",
"#| echo: false",
"knitr::include_graphics(current_xy_file)",
"```",
"",
":::",
"",
"::: {.column width='10%'}",
"",
":::",
"",
"::: {.column width='40%'}",
"",
"```{r eval=TRUE}",
"#| echo: false",
"knitr::include_graphics(current_umap_file)",
"```",
":::",
"",
":::",
"",
"",
""
), envir = environment(), quiet = TRUE)
})
cat(res, sep = '\n')
```
:::
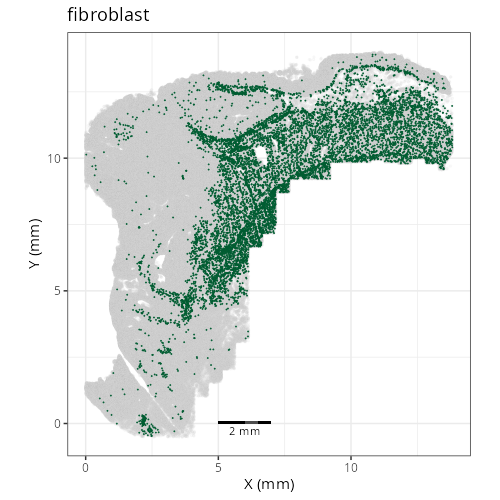
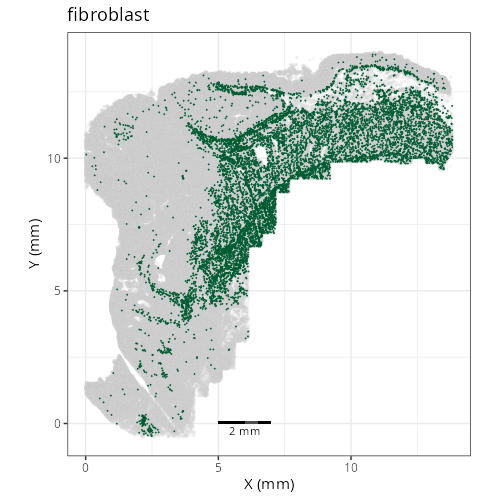
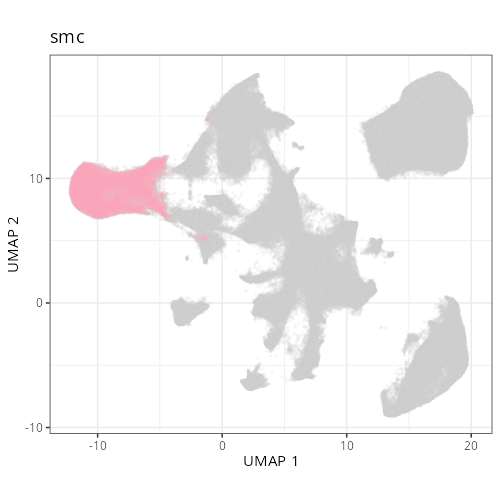
Note that within the SMC XY plots we can see an "outline" that appears to be the
muscularis mucosa.
## Integration with Spatial Domains {#sec-integration}
Recall in @sec-domains that we used `novae` to define spatial domains. Now that
we have cell type information, let's look at the composition of cell types
within each domain and assign a biological function to them.
Since we only need the metadata associated with the spatial domain results, we'll
only load the obs.
```{python}
#| label: I-1
#| eval: false
#| code-summary: "Python Code"
#| message: true
#| warning: false
#| code-fold: show
focal_domain_resolution = 'novae_domains_6'
if 'adata_domains' not in dir():
filename = os.path.join(r.analysis_dir, "anndata-3-novae.h5ad")
adata_domains = ad.read_h5ad(filename, backed='r')
meta_domains = adata_domains.obs.copy()
columns_to_copy = [focal_domain_resolution]
adata.obs[columns_to_copy] = meta_domains[columns_to_copy]
```
Create a contingency table for the domain and cell type assignments.
```{r}
#| label: I-2
#| eval: false
#| code-summary: "R Code"
#| message: true
#| warning: false
#| code-fold: show
focal_domain_resolution <- py$focal_domain_resolution
focal_celltype_resolution <- "celltype_fine"
df <- py$adata$obs %>% select(
all_of(focal_domain_resolution),
all_of(focal_celltype_resolution))
plot_data <- df %>%
group_by(.data[[focal_domain_resolution]],
.data[[focal_celltype_resolution]]) %>%
summarise(count = n()) %>%
mutate(proportion = count / sum(count))
# Create the geom_tile plot
p <- ggplot(plot_data, aes(x = .data[[focal_domain_resolution]],
y = .data[[focal_celltype_resolution]],
fill = proportion)) +
geom_tile(color = "white") +
geom_text(aes(label = paste0(round(100*proportion, 3), "%")), color = "black", size=2) +
scale_fill_gradient(low = "white", high = "dodgerblue") + # Set color scale
labs(y = "Cell Type", x = "Spatial Domain", fill = "Proportion") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
ggsave(p, filename = file.path(ct_dir, "contingency_table.png"),
width=5, height=7)
write.table(plot_data, "./tmp.csv", col.names=T, row.names=F, quote=F)
```
```{r}
#| label: fig-contingency-table
#| message: false
#| warning: false
#| echo: false
#| fig.width: 5
#| fig.height: 6
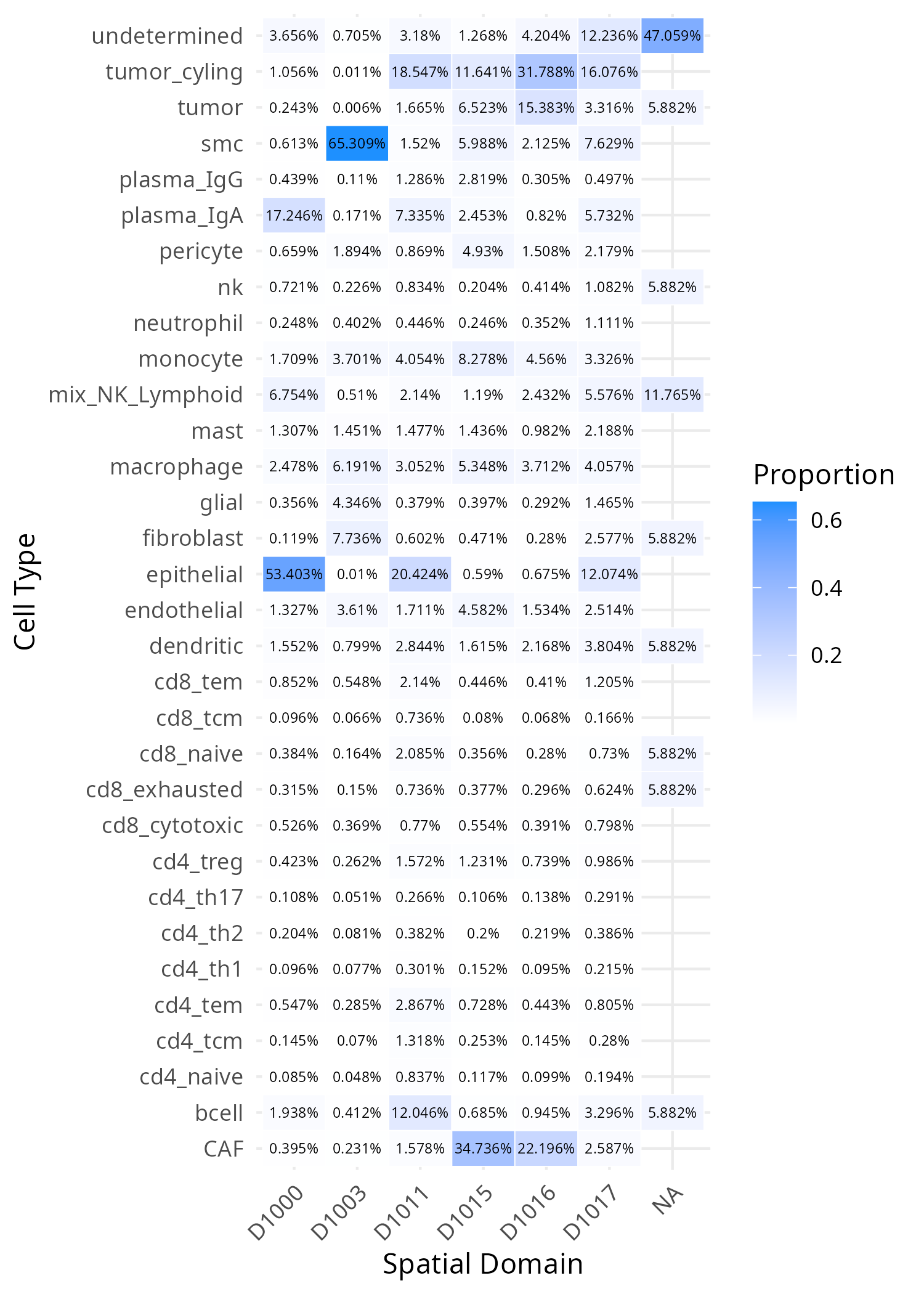
#| fig-cap: "The proportion of a given cell type found in each domain. For example, domain D1015 is comprised of 65.3% smooth muscle cells (SMCs)."
#| eval: true
render(file.path(analysis_asset_dir, "ct"), "contingency_table.png", ct_dir, overwrite=TRUE)
```
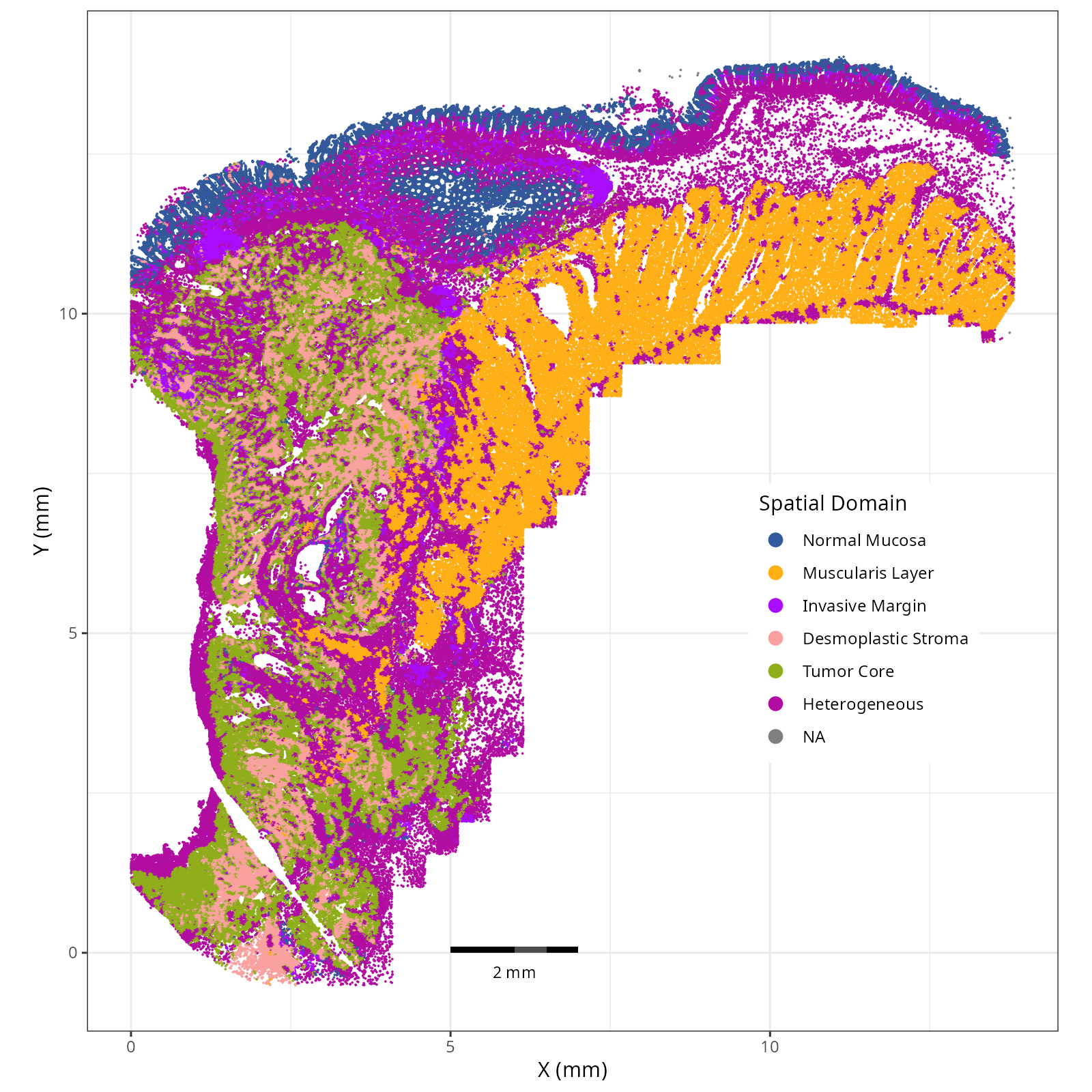
From this information and the spatial layout of domains we can assign a function
names @tbl-domains-definitions. Just looking at the composition, the differences
between three of the domains (D1011, D1015, and D1016) appear to have varying
degrees of tumor, cycling tumor, and CAFs. D1015 has relatively more CAFs so let's
call that a Desmoplastic Stroma domain and let's label D1016, which has the highest
concentration of tumor cells, the Tumor Core. D1011 has abundant tumor (cycling,
specifically) and also 12% B cell and 20% (normal) epithelial cells. Spatially,
D1011 occurs throughout lymphoid structures within the epithelium and around the
tumor region (invasive margin).
| Domain | Inferred Biological Identity | Top 3 Cell Types (Proportion) |
| :--- | :--- | :--- |
| **D1000** | **Normal Mucosa** | 1. epithelial (53.4%), 2. plasma_IgA (17.2%), 3. mix_NK_Lymphoid (6.8%) |
| **D1003** | **Muscularis Layer** | 1. smc (65.3%), 2. fibroblast (7.7%), 3. macrophage (6.2%) |
| **D1011** | **Invasive Margin (Immune-Rich)** | 1. epithelial (20.4%), 2. tumor_cyling (18.5%), 3. bcell (12.0%) |
| **D1015** | **Desmoplastic Stroma** | 1. CAF (34.7%), 2. tumor_cyling (11.6%), 3. monocyte (8.3%) |
| **D1016** | **Tumor Core** | 1. tumor_cyling (31.8%), 2. CAF (22.2%), 3. tumor (15.4%) |
| **D1017** | **Heterogeneous** | 1. tumor_cyling (16.1%), 2. undetermined (12.2%), 3. epithelial (12.1%) |
: Functional names associated with each `novae` domain. {#tbl-domains-definitions}
Let's add a new column in the metadata with these values.
```{python}
#| label: I-3
#| eval: false
#| code-summary: "Python Code"
#| message: true
#| warning: false
#| code-fold: show
domain_map = {
'D1000': 'Normal Mucosa',
'D1003': 'Muscularis Layer',
'D1011': 'Invasive Margin',
'D1015': 'Desmoplastic Stroma',
'D1016': 'Tumor Core',
'D1017': 'Heterogeneous'
}
adata.obs['annotated_domain'] = adata.obs[focal_domain_resolution].map(domain_map)
filename = os.path.join(r.analysis_dir, "anndata-7-domain_annotations.h5ad")
adata.write_h5ad(
filename,
compression=hdf5plugin.FILTERS["zstd"],
compression_opts=hdf5plugin.Zstd(clevel=5).filter_options
)
```
And while were at it, let's plot the domains in XY space using `plotDots` with
the new labels.
```{r}
#| label: I-4
#| eval: false
#| code-summary: "R Code"
#| message: true
#| warning: false
#| code-fold: show
domain_colors <- results_list[['novae_model_1_domain_colors']]
names(domain_colors) <- results_list[['novae_model_1_domain_names']]
domain_map <- py$domain_map
domain_map <- data.frame('domain_id' = names(domain_map),
'domain_description'=unlist(domain_map))
domain_colors <- domain_colors[match(domain_map$domain_id, names(domain_colors))]
domain_map$domain_color <- domain_colors
results_list[['domain_colors']] <- domain_map
saveRDS(results_list, results_list_file)
colors_use <- domain_map$domain_color
names(colors_use) <- domain_map$domain_description
group_prefix <- "annotated_domain"
plotDots(py$adata, color_by='annotated_domain',
plot_global = TRUE,
facet_by_group = FALSE,
additional_plot_parameters = list(
geom_point_params = list(
size=0.001
),
scale_bar_params = list(
location = c(5, 0),
width = 2,
n = 3,
height = 0.1,
scale_colors = c("black", "grey30"),
label_nudge_y = -0.3
),
directory = ct_assets_dir,
fileType = "png",
dpi = 200,
width = 8,
height = 8,
prefix=group_prefix
),
additional_ggplot_layers = list(
theme_bw(),
xlab("X (mm)"),
ylab("Y (mm)"),
coord_fixed(),
scale_color_manual(values = colors_use),
theme(legend.position = c(0.8, 0.4)),
guides(color = guide_legend(
title="Spatial Domain",
override.aes = list(size = 3) ) )
)
)
```
```{r}
#| label: fig-xy-domains
#| message: false
#| warning: false
#| echo: false
#| fig.width: 6
#| fig.height: 5
#| fig-cap: "XY with spatial domains."
#| eval: true
knitr::include_graphics(file.path(analysis_asset_dir, "ct", "annotated_domain__spatial__S0__plot.png"))
```
## Conclusion
We have now mapped the cellular landscape of our tissue. By combining broad
unsupervised clustering and the specialized
HieraType algorithm, we have moved from a matrix of counts to a spatially
annotated map. Then we used this information to create functional names to
the domains that we found in @sec-domains.
It is important to recognize that this cell typing workflow is a foundational
draft. Depending on your specific research questions, such as defining specialized
epithelial cell types or mapping complex stromal niches, more advanced methods may be
required.
With our cells now labeled with a type and a domain, we are ready to ask
"tertiary" questions. At the moment, this includes an analysis on pathway
enrichment but more analysis chapters are in the works.