---
subtitle: Converting files
author:
- name: Evelyn Metzger
orcid: 0000-0002-4074-9003
affiliations:
- ref: bsb
- ref: eveilyeverafter
execute:
eval: false
freeze: auto
message: true
warning: false
self-contained: false
code-fold: false
code-tools: true
code-annotations: hover
format: live-html
engine: knitr
freeze: true
---
{{< include ./_extensions/r-wasm/live/_knitr.qmd >}}
# Data Wrangling {#sec-data-wrangle}
```{r}
#| label: Preamble
#| eval: true
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
source("./preamble.R")
reticulate::source_python("./preamble.py")
analysis_dir <- file.path(getwd(), "analysis_results")
input_dir <- file.path(analysis_dir, "input_files")
output_dir <- file.path(analysis_dir, "output_files")
analysis_asset_dir <- "./assets/analysis_results" # <1>
qc_dir <- file.path(output_dir, "qc")
if(!dir.exists(qc_dir)){
dir.create(qc_dir, recursive = TRUE)
}
results_list_file = file.path(analysis_dir, "results_list.rds")
if(!file.exists(results_list_file)){
results_list <- list()
saveRDS(results_list, results_list_file)
} else {
results_list <- readRDS(results_list_file)
}
```
1. This puts select images into the projects assets directory to enhance portability.
No data analysis journey can truly begin without first converting _something_ and our task
for this chapter is simple: take the [flat files](https://nanostring-biostats.github.io/CosMx-Analysis-Scratch-Space/posts/flat-file-exports/flat-files-compare.html) that are exported from AtoMx SIP
into a usable format. That format is primarily python's anndata format but, interestingly
enough, we'll take advantage of R's `data.table` @barrett2025 package to
efficiently read in the expression matrix file. I find this is much faster than
reading in the expression matrix into pandas.
::: {.column-margin}
::: {.otherapproachesbox title="Alternative Approach"}
While this chapter focusing on converting the flat files into anndata format,
it skips over other types of data that we can convert and use -- such as the imaging
data. Both Squidpy @squidpy and SpatialData @spatialdata offer ways to include composite images[^these are single
images per FOV that combine the various IF channels together] into anndata objects.
One limitation of these approaches -- at the time of writing -- is that they treat
each FOV separately making it difficult to view results when you working with several
hundred FOVs. For a tutorial on how to visualize the per-channel images across the entire
sample with Napari, see this
[Scratch Space Blog](https://nanostring-biostats.github.io/CosMx-Analysis-Scratch-Space/posts/napari-cosmx-intro/).
<br>
In addition to processing and analyzing with python, R offers ways to analyze CosMx
SMI data using packages like `Seurat` @seurat. For an example on how to use Seurat for visualization,
see Claire William's [Blog post](https://nanostring-biostats.github.io/CosMx-Analysis-Scratch-Space/posts/seurat-cosmx-basics/) and other vignettes.
:::
:::
Split the expression data into targets, negatives, and system controls. Read
in the metadata and add columns that represent the slide position in micrometers.
```{r}
#| label: convert-data-r
#| eval: true
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
tmp_dir <- "./tmp_dir"
dir.create(tmp_dir, showWarnings = FALSE) # <1>
flat_file_dir <- file.path(input_dir, "Run_b8806732_S2_Colon")
expression_dt <- data.table::fread(
file.path(flat_file_dir, "S0_exprMat_file.csv.gz"),
tmpdir = tmp_dir)
colnames(expression_dt)[
which(colnames(expression_dt) == "cell_ID")] <- 'cell'
expression_dt <- expression_dt %>%
arrange(fov, cell)
expression_dt <- filter(expression_dt, cell!=0) # <2>
expression_dt <- expression_dt %>%
mutate('cell_ID' = paste0("c_1_", fov, "_", cell))
cell_ids_vector_r <- expression_dt$cell_ID
all_feature_names <- setdiff(
names(expression_dt), c("fov", "cell_ID", "cell")) # <3>
neg_indices_r <- which(
startsWith(all_feature_names, "Negative") |
startsWith(all_feature_names, "NegPr"))
sys_indices_r <- which(
startsWith(all_feature_names, "System") |
startsWith(all_feature_names, "False"))
gene_indices_r <- which(!startsWith(all_feature_names, "Negative") &
!startsWith(all_feature_names, "System") &
!startsWith(all_feature_names, "NegPr") &
!startsWith(all_feature_names, "False"))
neg_probe_names_vector_r <- all_feature_names[neg_indices_r]
sys_probe_names_vector_r <- all_feature_names[sys_indices_r]
gene_probe_cols <- all_feature_names[gene_indices_r]
neg_mat_r <- as.matrix(expression_dt[, ..neg_probe_names_vector_r])
sys_mat_r <- as.matrix(expression_dt[, ..sys_probe_names_vector_r])
gene_mat_r <- as.matrix(expression_dt[, ..gene_probe_cols])
meta <- data.table::fread(
file.path(flat_file_dir, "S0_metadata_file.csv.gz"))
meta$tmp <- meta$cell_ID
meta$cell_ID <- meta$cell_id
meta$cell_id <- meta$tmp
meta$tmp <- NULL
meta <- meta %>% arrange(fov, cell_id)
um_per_px = 0.1203 # <4>
meta$x_slide_mm <- um_per_px*meta$CenterX_global_px/1e3
meta$y_slide_mm <- um_per_px*meta$CenterY_global_px/1e3
if(nrow(meta) != nrow(gene_mat_r)){
stop("Error. Check file inputs.")
}
fov_locations <- data.table::fread(
file.path(flat_file_dir, "S0_fov_positions_file.csv.gz")) # <5>
nanoparquet::write_parquet(
fov_locations %>% select(FOV, x_global_mm, y_global_mm),
file="./assets/interactives/fov_locations.parquet")
```
1. My EC2 instance only has 100 GBs of storage. Setting it to a mounted drive without such restriction avoids low disk space warnings or complications.
2. We want to exclude 'cell 0' here. This is a special identifier for transcripts
not assigned within a cell boundary.
3. This includes targets, negatives, and system controls
4. Conversion: 0.1203 µm per pixel
5. Used in a qc processing section
While the matrix and metadata information do not contain cell segmentation detail,
it's always a good idea to plot the cells in space to ensure they match
our expected orientation.
```{r}
#| label: xy-cells-initial
#| eval: true
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "R code"
p <- ggplot(data=meta) +
geom_point(
aes(x=x_slide_mm, y=y_slide_mm),
size=0.001, alpha=0.1) +
coord_fixed() + theme_bw()
ggsave(filename = file.path(qc_dir, "xy_cells_initial_lowres.png"), p,
width=8, height=8, dpi=80, type = 'cairo') # <1>
ggsave(filename = file.path(qc_dir, "xy_cells_initial_highres.png"), p,
width=8, height=8, dpi=400, type = 'cairo')
```
1. I tend to plot lower-resolution versions of pngs to make the Quarto book light weight.
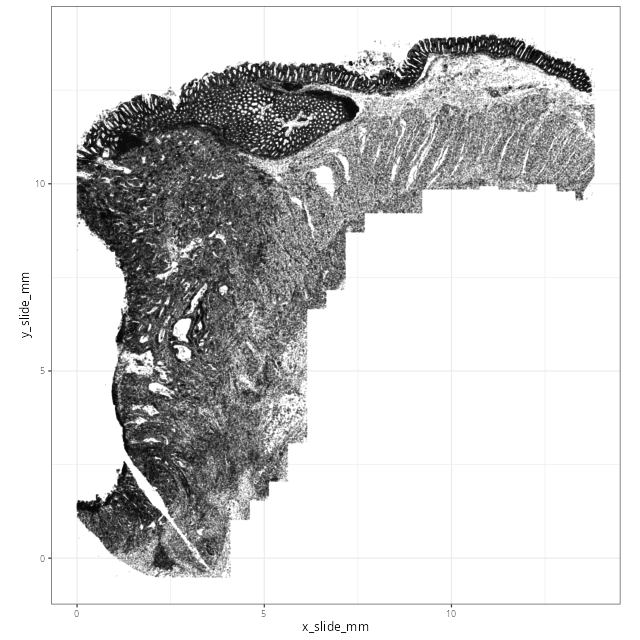
@fig-xy-cells-initial confirms the expected orientation and alignment from the
flat files.
```{r}
#| label: fig-xy-cells-initial
#| eval: true
#| echo: false
#| code-summary: "R Code"
#| fig-cap: "Spatial oreientation of cells."
#| fig-align: center
#| out-width: "70%"
render(file.path(analysis_asset_dir, 'qc'), "xy_cells_initial_lowres.png", source_parent_folder=qc_dir)
```
With that confirmation, create the initial anndata object. Specifically, we'll start with
dense^[I generally avoid handing off sparse matrices from R to python and _vice versa_ and
since the data were dense to begin with (_i.e._, it was a CSV file), we'll make it sparse in with python.]
matrices of expression, system controls (_i.e._, "false codes"), and negative probes,
and add these into the `X` elment and into `obsm`, respectively. We'll place the
metadata into the `obs` and we'll also create a matrix of (X, Y) coordinates into
their own `obsm`.
When analyzing interactively
I switch between R and python blocks explicitly with `reticulate::repl_python()` to
enter python and type `exit()` to return back to R.
```{python}
#| label: create-anndata
#| eval: true
#| echo: true
#| message: false
#| code-fold: show
#| code-summary: "Python code"
filename = os.path.join(r.analysis_dir, "anndata-0-initial.h5ad")
if not os.path.exists(filename):
# 1 Initial construction
adata = ad.AnnData(
X=r.gene_mat_r,
obsm={'counts_neg': sp.sparse.csr_matrix(r.neg_mat_r), 'counts_sys': sp.sparse.csr_matrix(r.sys_mat_r)},
obs=r.meta,
var=pd.DataFrame(index=r.gene_probe_cols),
)
adata.X = sp.sparse.csr_matrix(adata.X)
adata.uns['neg_col_names'] = r.neg_probe_names_vector_r
adata.uns['sys_col_names'] = r.sys_probe_names_vector_r
# 2. add coordinates in the 'spatial' obsm
coordinates = adata.obs[['x_slide_mm', 'y_slide_mm']].to_numpy() # <1>
adata.obsm['spatial'] = coordinates
# 3. Save object to disk
adata.write_h5ad(
filename,
compression=hdf5plugin.FILTERS["zstd"],
compression_opts=hdf5plugin.Zstd(clevel=5).filter_options
)
```
1. Some packages expect the xy coordinates as a numpy array in obsm.
And that's it! Our data have been converted into anndata format and now we're
ready to assess the quality.