Terminal
pip install numpy scipy opencv-python scikit-imageLidan Wu 
The ability to separate cells based on their relationship to nearby marker protein staining is a desired feature in many biological studies. One such use case would be to study the difference between neurons immediately next to neurofibrillary tangles and neurons that are a little further away. Another example would be to study the immune cells that are at different stages of engulfing microbes. This post introduces a pipeline for automatically generating GeoMx® Digital Spatial Profiler (DSP)-ready binary masks in batch for marker-based single-cell application.
Like other items in our Analysis Scratch Space, the usual caveats and license apply.
The pipeline is a python script Generate_scMask_given_query.py that runs as a command line and you can downloaded from _code/GeoMx-scMask-generation folder in this repository. By passing the file path to image folder to the script, the pipeline would take GeoMx® morphology images and generate binary masks for negative-stained cells and cells connecting to positive-stained regions, respectively.
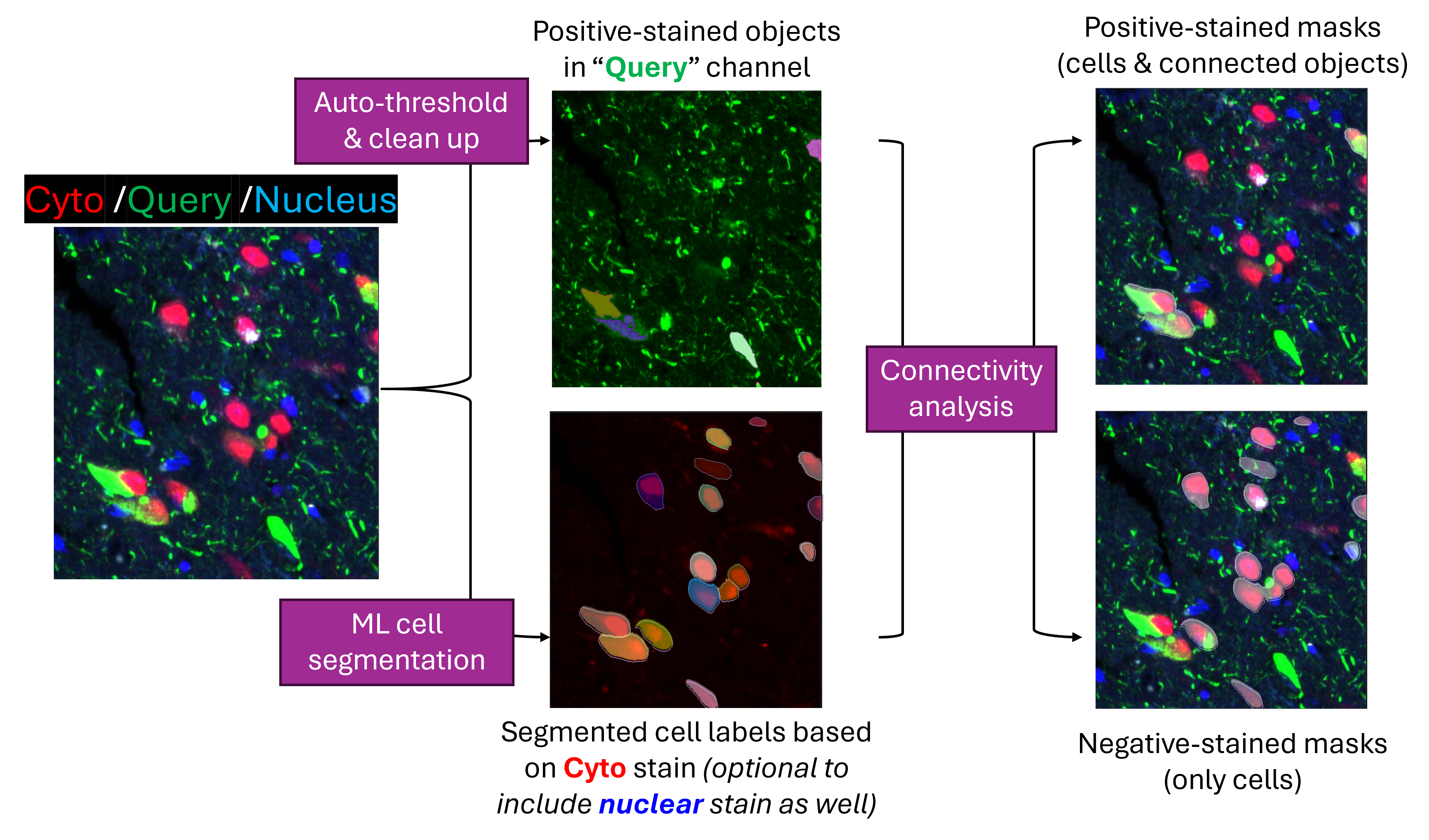
2chan_res (or 3chan_res if including nuclear stain as 3rd channel) sub folder of the user-designated output root folder.cellLabels sub folder within the step (1) result folder.min_positive_area.
negative_cell_masks: binary masks for cells that do NOT have valid overlapping with any of positive-stained object.
min_intersect_area.positive_ROI_masks: binary masks for cells with valid overlapping event AND their connected positive-stained objects.Once complete, user can upload these binary masks back to GeoMx® DSP instrument for the corresponding ROI selection and initiate the sample collection.
You can use ImageJ or similar viewer to inspect and modify the outcomes of both cell segmentation and binary mask generation. We recommend cellpose GUI for visualization and modification on cell segmentation outcomes.
To use the modified cell label images which must be under same names and folder structure as original outputs, simply rerunning the pipeline script with same configuration would skip the step (1) and (2) above but instead perform identification of positive-stained objects and binary mask generation using the modified cell label images.
The pipeline script uses cellpose python package for cell segmentation. Please refer to cellpose guide on installation instruction for best usage, especially if you want to enable GPU for faster processing.
In addition, you would also need the following python packages in your environment. Below shows how to install them in terminal via pip.
Terminal
pip install numpy scipy opencv-python scikit-imageThe pipeline script in use, Generate_scMask_given_query.py, assumes you have multi-channel single-slice images in one folder. Those images should be downloaded for the ROIs you choose in GeoMx® study. The required inputs for the pipeline include:
in_dir: the absolute path to your GeoMx® DSP morphology image folder.cyto_chan, query_chan, nuc_chan: channel index for cytoplasm, query marker and nucleus stain, respectively.
--cyto_chan 4 with the script.nuc_chan is set to 0 such that it would not be included in cell segmentation downstream. But if you would like to use stains for both cytoplasm and nucleus for cell segmentation, you can pass the corresponding channel index of nucleus stain to the script.cell_diameter: median cell diameter in pixel unit within your images, default to 30. For best cell segmentation results, it’s recommended to set this parameter based on your sample.To use the pipeline, run python Generate_scMask_given_query.py and specify parameters as desired.
For instance to run on a folder with images where the 4th and 3rd channels of each image are cytoplasm and query marker stain, respectively while defining positive-stained objects (in query marker channel) to have at least 500 squared pixel area (using default values for all other parameters):
Command Line Interface (CLI) example to run in Terminal
python Generate_scMask_given_query.py --in_dir imageFolder --cyto_chan 4 --query_chan 3 --out_dir outputFolder --min_positive_area 500The resulting output folder structure looks like this:
outputFolder
├── 20240612_pipeline_run_log.txt
│
├── 2chan_res
│ ├── 001.tiff
│ ├── 002.tiff
│ └──cellLabels
│ ├── 001_cp_masks.tif
│ └── 002_cp_masks.tif
│
├── negative_cell_masks
│ ├── 001_neg_2.tif
│ └── 002_neg_2.tif
│
└── positive_ROI_masks
├── 001_pos_1.tif
└── 002_pos_1.tifThere are other additional arguments providing fine control of different steps of the pipeline. Please use -h argument to see a full list of options allowed by the pipeline script.
Command Line Parameters
usage: Generate_scMask_given_query.py [-h] [--in_dir IN_DIR] [--out_dir OUT_DIR] [--cyto_chan CYTO_CHAN]
[--query_chan QUERY_CHAN] [--nuc_chan NUC_CHAN]
[--cell_seg_model CELL_SEG_MODEL] [--cell_diameter CELL_DIAMETER]
[--min_cell_area MIN_CELL_AREA] [--thresh_method THRESH_METHOD]
[--fill_holes] [--min_positive_area MIN_POSITIVE_AREA]
[--min_intersect_area MIN_INTERSECT_AREA] [--clean_export]cyto3
triangle or otsu; Default: triangle
True, use 0.1x of input cell diameter as kernel size and output roundish borders; Default: False
False